



No CrossRef data available.
Article contents
Meifuite, A New Ferrous Phyllosilicate Mineral With Modulated Tetrahedral Sheets Similar to Minnesotaite
Published online by Cambridge University Press: 01 January 2024
Abstract
A new ferrous phyllosilicate, meifuite, has been discovered in the Yinachang Fe-Cu-REE (rare-earth element) deposit in China. The structural formula, calculated using averaged electron probe microanalysis (EPMA) results, is K0.72Na0.20(Fe5.56Mg0.31Mn0.13)Σ6.00(Si6.95Al1.04)Σ7.99O18.84(OH)4.84 Cl1.33, with an ideal formula of KFe6(AlSi7)O19(OH)4Cl2. The structure of meifuite has a P1¯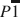 space group symmetry, with unit-cell parameters of a = 22.7773(13) Å, b = 9.5553(5) Å, c =14.3282(8) Å, α = 99.258(4)°, β = 136.750(3)°, γ = 89.899(4)°, Z = 2, and V = 2077.9(2) Å3. Meifuite has a strip-modulated 2:1 layer (T–O–T) structure similar to that of minnesotaite. About 1/8 of the tetrahedra in the T sheet are occupied by Al instead of Si, and the interlayer cavities are partially occupied by K and Na. Some of the OH sites in the octahedral sheet in the layer structure are fully or partially substituted by Cl, which is apparently the primary reason for the meifuite structure being more stable than stilpnomelane, the most common ferrous layer silicate mineral found at similar temperature and pressure conditions. An updated, more accurate structure model of minnesotaite is also provided for comparison with the meifuite structure. The mineral is named after Meifu Zhou in honor of his outstanding contributions to the field of economic geology.
space group symmetry, with unit-cell parameters of a = 22.7773(13) Å, b = 9.5553(5) Å, c =14.3282(8) Å, α = 99.258(4)°, β = 136.750(3)°, γ = 89.899(4)°, Z = 2, and V = 2077.9(2) Å3. Meifuite has a strip-modulated 2:1 layer (T–O–T) structure similar to that of minnesotaite. About 1/8 of the tetrahedra in the T sheet are occupied by Al instead of Si, and the interlayer cavities are partially occupied by K and Na. Some of the OH sites in the octahedral sheet in the layer structure are fully or partially substituted by Cl, which is apparently the primary reason for the meifuite structure being more stable than stilpnomelane, the most common ferrous layer silicate mineral found at similar temperature and pressure conditions. An updated, more accurate structure model of minnesotaite is also provided for comparison with the meifuite structure. The mineral is named after Meifu Zhou in honor of his outstanding contributions to the field of economic geology.
- Type
- Article
- Information
- Copyright
- Copyright © Clay Minerals Society 2021
References
Ahn, J. H., & Buseck, P. R. (1989). Microstructures and tetrahedral strip-width order and disorder in Fe-rich minnesotaites. American Mineralogist, 74, 384–393.Google Scholar
Blake, R. L. (1965). Iron phyllosilicates of the Cuyuna district in Minnesota. American Mineralogist, 50, 148–169.Google Scholar
Blöchl, P. E. (1994). Projector augmented-wave method. Physical Review B, 50, 17953–17979.Google Scholar
Brigatti, M.F. & Guggenheim, S. (2002). Mica crystal chemistry and the influence of pressure, temperature, and solid solution on atomistic models. Pp. 1–97 in: Micas: Crystal Chemistry and Metamorphic Petrology (Mottana, A., Sassi, F.P., and Thompson, James B. Jr, editors). Reviews in Mineralogy and Geochemistry, 46, Mineralogical Society of America, Chantilly, VA, USA.Google Scholar
Brown, E.H. (1971). Phase relations of biotite and stilpnomelane in the greenschist facies. Contributions to Mineralogy and Petrology, 31, 275–299.Google Scholar
Eggleton, R. A. (1972). The crystal structure of stilpnomelane. Part II. The full cell. Mineralogical Magazine, 38, 693–711.Google Scholar
Eggleton, R. A., & Bailey, S. W. (1964). The crystal structure of stilpnomelane Part I. The Subcell. Clays and Clay Minerals, 13, 49–63.Google Scholar
Eggleton, R. A., & Chappell, B. W. (1978). The crystal structure of stilpnomelane. Part III: Chemistry and physical properties. Mineralogical Magazine, 42, 361–368.Google Scholar
Eggleton, R. A., & Guggenheim, S. (1986). A re-examination of the structure of ganophyllite. Mineralogical Magazine, 50, 307–315.Google Scholar
Giese, R. F. (1979). Hydroxyl orientations in 2: 1 phyllosilicates. Clays and Clay Minerals, 27, 213–223.Google Scholar
Guggenheim, S., & Bailey, S. W. (1982). The superlattice of minnesotaite. The Canadian Mineralogist, 20, 579–584.Google Scholar
Guggenheim, S., & Eggleton, R. A. (1986a). Cation exchange in ganophyllite. Mineralogical Magazine, 50, 517–520.Google Scholar
Guggenheim, S., & Eggleton, R. A. (1986b). Structural modulations in iron-rich and magnesium-rich minnesotaite. The Canadian Mineralogist, 24, 479–497.Google Scholar
Guggenheim, S., & Eggleton, R. A. (1987). Modulated 2: 1 layer silicates: review, systematics, and predictions. American Mineralogist, 72, 724–738.Google Scholar
Guggenheim, S., & Eggleton, R. A. (1994). A comparison of the structures and geometric stabilities of stilpnomelane and parsettensite: A distance least-squares (DLS) study. American Mineralogist, 79, 438–442.Google Scholar
Guggenheim, S., & Eggleton, R. A. (1998). Modulated crystal structures of greenalite and caryopilite: A system with long-range, in-plane structural disorder in the tetrahedra sheet. The Canadian Mineralogist, 36, 163–179.Google Scholar
Hazen, R. M., & Burnham, C.W. (1973). The crystal structures of one-layer phlogopite and annite. American Mineralogist, 58, 889–900.Google Scholar
Jefferson, D. A. (1976). Stacking disorder and polytypism in zussmanite. American Mineralogist, 61, 470–483.Google Scholar
Jin, S., Li, X., & Xu, H. (2020). Meifuite, IMA 2019-101. CNMNC Newsletter No. 54. Mineralogical Magazine, 84. https://doi.org/10.1180/mgm.2020.21Google Scholar
Jin, S., Xu, H., Lee, S., & Fu, P. (2018). Jinshajiangite: structure, twinning and pseudo-symmetry. Acta Crystallographica Section B, 74, 325–336.Google Scholar
Klein, C. (1974). Greenalite, stilpnomelane, minnesotaite, crocidolite and carbonates in a very low-grade metamorphic Precambrian iron formation. The Canadian Mineralogist, 12, 475–498.Google Scholar
Kresse, G., & Furthmülller, J. (1996). Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Physical Review B, 54, 11169–11186.Google Scholar
Kresse, G., & Joubert, D. (1999). From ultrasoft pseudopotentials to the projector augmented-wave method. Physical Review B, 59, 1758–1775.Google Scholar
Leissner, L. (2014). Crystal chemistry of amphiboles studied by Raman spectroscopy. Mineralogisch-Petrographisches Institut-Universität Hamburg, Hamburg, Germany, 45 pp.Google Scholar
Li, X., Zhao, X., Zhou, M.-F., Chen, W. T., & Cho, Z. (2015). Fluid Inclusion and Isotopic Constraints on the Origin of the Paleoproterozoic Yinachang Fe-Cu-(REE) Deposit, Southwest China. Economic Geology, 110, 1339–1369.Google Scholar
Loewenstein, W. (1954). The distribution of aluminum in the tetrahedra of silicates and aluminates. American Mineralogist, 39, 92–96.Google Scholar
Lopes-Vieira, A., & Zussman, J. (1969). Further detail on the crystal structure of zussmanite. Mineralogical Magazine, 37, 49–60.Google Scholar
Miyano, T. (1982). Stilpnomelane, iron-rich mica, K-feldspar and hornblende in banded iron-formation assemblages of the Dales Gorge Member, Hamersley Group, Western Australia. The Canadian Mineralogist, 20, 189–202.Google Scholar
Miyano, T., & Klein, C. (1989). Phase equilibria in the system K2O-FeO-MgO-Al2O3-SiO2-H2O-CO2 and the stability limit of stilpnomelane in metamorphosed Precambrian iron-formations. Contributions to Mineralogy and Petrology, 102, 478–491.Google Scholar
Momma, K., & Izumi, F. (2011). VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. Journal of Applied Crystallography, 44, 1272–1276.Google Scholar
Perdew, J. P., Burke, K., & Ernzerhof, M. (1996). Generalized gradient approximation made simple. Physical Review Letters, 77, 3865–3868.Google Scholar
Petříček, V., Dušek, M., & Palatinus, L. (2014). Crystallographic computing system JANA2006: General features. Zeitschrift für Kristallographie-Crystalline Materials, 229, 345–352.Google Scholar
Volfinger, M., Robert, J.-L., Vielzeuf, D., & Neiva, A. M. R. (1985). Structural control of the chlorine content of OH-bearing silicates (micas and amphiboles). Geochimica et Cosmochimica Acta, 49, 37–48.Google Scholar
Wang, L., Maxisch, T., & Ceder, G. (2006). Oxidation energies of transition metal oxides within the GGA+U framework. Physical Review B, 73, 195107.Google Scholar
Wang, A., Freeman, J. J., & Jolliff, B. L. (2015). Understanding the Raman spectral features of phyllosilicates. Journal of Raman Spectroscopy, 46, 829–845.Google Scholar

Jin et al. supplementary material

File
240.8 KB