


Introduction
Lambert–Eaton syndrome (LES) is a rare autoimmune disorder clinically characterized by weakness, depressed or absent reflexes and autonomic dysfunction. LES is believed to be triggered in some cases by the presence of a tumour, most often small-cell lung cancer (SCLC), or in others (non-paraneoplastic LES) by an unknown autoimmune dysregulation that provokes a cross-reactive autoantibody response. Autoantibodies targeted to P/Q-type voltage-gated calcium channels (VGCCAb) are present in about 90% of LES patients. These antibodies impair cholinergic release at the skeletal neuromuscular junction and the autonomic synapses. Although this syndrome has historically been referred to as Lambert–Eaton “myasthenic” syndrome, the clinical manifestations are not restricted to just myasthenic symptoms, and non-muscle symptoms are often misleading and can lead to diagnostic error. LES is a disabling but rarely life-threatening disease. Severe respiratory dysfunction and autonomic instability are rare, but it is critical to recognize these conditions because they may contribute to worsening disability and impaired quality of life. The nature of LES onset is variable, as it may precede the diagnosis of a malignant or autoimmune disease, occur late in the illness or herald tumour recurrence.Reference Beckles, Spiro, Colice and Rudd 1 Therefore, early diagnosis and treatment may improve clinical outcomes.Reference de Beukelaar and Sillevis Smitt 2
This review highlights the most common diagnostic pitfalls in the clinical approach to LES and focuses on the usefulness of autonomic and respiratory testing. A literature search of electronic databases (PubMed and Web of Science) for articles published in English up until January of 2016 was performed using the keywords “Lambert”, “Eaton”, and “myasthenic”. Papers from the reference list of these publications were screened for descriptions of additional cases. Additionally, we report a series of three cases in which a neurophysiologic assessment was conducted for various parts of the nervous system, including autonomic functioning and the respiratory system.
Clinical Approach to LES
Clinical Presentation, Muscle Weakness and Progression
LES is frequently misdiagnosed given its non-specific and fluctuating symptoms, which often appear within a prolonged time frame. The clinical course of LES is generally progressive, with less fluctuation and a lower spontaneous remission rate than myasthenia gravis (MG).Reference Harper and Lennon 3 - Reference Bromberg, Albers and McCune 5 In paraneoplastic LES patients (P-LES), symptoms and findings spread far more rapidly and can be initially attributed to cachexia and/or effects of cancer or its treatment.Reference Wirtz, Wintzen and Verschuuren 6
LES presents with fatigue, proximal weakness, particularly in the lower limbs, and unanticipated falls. Although weakness usually progresses insidiously, it may develop following infectious process or flare-up of a previously diagnosed autoimmune disease.Reference Bromberg, Albers and McCune 5 , Reference Juel and Sanders 7 Clinicians should be aware that muscle weakness on examination in LES patients may be minimal and out of proportion to their complaints, and thus avoid dismissing symptoms as psychogenic. Ache, stiffness and tenderness to palpation, aggravated by exercise, are common complaints in back and affected muscle groups.Reference Wirtz, Smallegange, Wintzen and Verschuuren 8 , Reference Simon, Herbison and Levy 9 These symptoms, when prominent, can mislead the diagnosis of neurogenic claudication associated with canal spinal stenosis.
As the disease progresses, most LES patients eventually develop cranial weakness, which may lead to an erroneous diagnosis of MG. The initial distribution of symptoms, the degree and severity of weakness, and the subsequent progression are distinguishing clinical clues between both entities (Table 1). For more information on clinical presentation and muscle weakness, see the review by Verschuuren et al. (2014).Reference Verschuuren, Titulaer and Maddison 10
Table 1 Main Features in the Differential Diagnosis Between Lambert–Eaton Syndrome and Myasthenia Gravis
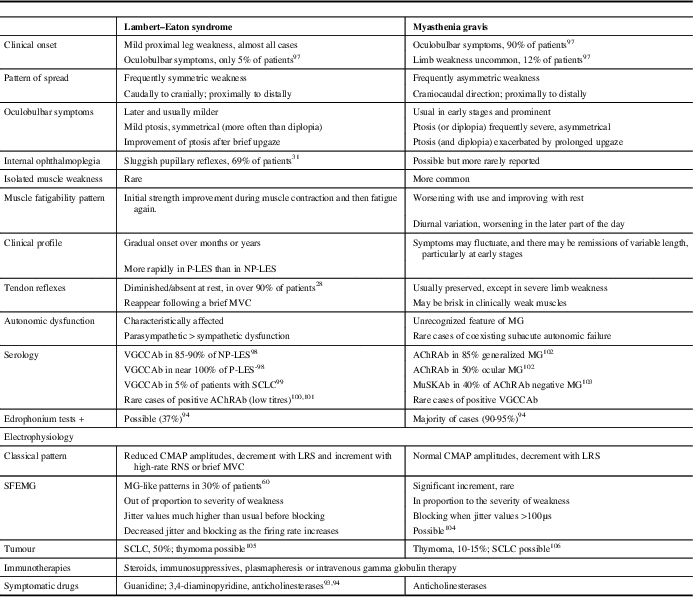
P-LES=paraneoplastic LES; NP-LES=non-paraneoplastic LES; MVC=maximal voluntary contraction; MG=myasthenia gravis; VGCCAb=antibodies against voltage-gated calcium channels; AChRAb=acetylcholine receptor antibodies; MuSKAb=muscle-specific kinase antibodies; LRS=low-rate stimulation; RNS=repetitive nerve stimulation; CMAP=compound muscle action potential; SFEMG=single-fibre electromyography.
Associated autoimmune disorders can occur in up to 33% of non-paraneoplastic LES (NP-LES) patients.Reference Tim, Massey and Sanders 11 Clinicians should be aware that manifestations of coexisting organ-specific autoimmune disorders (e.g., hyperthyroidism or hypothyroidism, pernicious anaemia) are common in these patients and can be misleading.Reference Lennon, Lambert, Whittingham and Fairbanks 12
The diagnosis of LES may be overlooked when symptoms and signs of multiple paraneoplastic disorders coexist, particularly in the setting of subacute cerebellar ataxia or severe peripheral neuropathy.Reference Nagashima, Mizutani, Kawahara, Maguchi, Terayama and Shinohara 13 - Reference Kalra, Gozzard, Jacob, Leonard and Maddison 15 The sensory examination is normal in most LES patients unless a coexistent peripheral neuropathy, paraneoplastic sensory neuronopathy, myelopathy or encephalopathy is present.Reference Kleopa, Teener, Scherer, Galetta and Bird 16
Respiratory Muscle Involvement in LES
Relatively frequent reports of respiratory muscle involvement in LES suggest that this condition is more common than is generally recognized, and LES may often go underdiagnosed in patients with respiratory failure of undetermined cause.Reference Bolton 17
The weakness in LES is rarely a life-threatening condition unless vital muscles are severely affected. Respiratory muscle involvement is usually mild, with an unspecific restrictive pattern on spirometry. Symptoms of dyspnea are unusual and are commonly attributed to the underlying lung disease in P-LES patients.Reference Meriggioli 18
Rapidly progressing respiratory failure without other prominent symptoms is infrequent in LES,Reference Nicolle, Stewart, Remtulla, Chen and Bolton 19 compared with MG, and may be precipitated by intercurrent pulmonary pathology or drugs that impair neuromuscular transmission.Reference Weingarten, Araka, Mogensen, Sorenson, Marienau and Watson 20 - Reference Tseng, Claussen and Oh 23 Respiratory failure with prolonged mechanical ventilation following anaesthesia may be a presenting feature, and reversal with anticholinesterases may be incomplete.Reference Telford and Hollway 24 An unduly prolonged paralysis can also occur after using neuromuscular blockers.Reference Small, Ali, Lennon, Brown, Carr and de Armendi 25 A high index of suspicion is required to make the diagnosis in this setting.
Because of the association of LES with lung cancer, the presence of unexplained dyspnea should arouse suspicion of diaphragmatic muscle weakness secondary to phrenic nerve injury. SCLC is considered the second most common cause of diaphragmatic paralysis because of its highly aggressive regional spread, including invasion of the phrenic nerves.Reference Bolton 26 The left phrenic nerve is more commonly affected than the right, probably because of greater proximity to the lymph nodes of the aortopulmonary window. The diagnosis of this condition is indicative of locally advanced disease, which is generally not reversible.Reference Wozniak and Gadgeel 27
Autonomic Dysfunction in LES
Symptoms or signs of autonomic dysfunction have been found in up to 80% of LES patients and may be the presenting symptom in 6% of patients, preceding muscle weakness onset by years.Reference O’Neill, Murray and Newsom-Davis 28 , Reference O’Suilleabhain, Low and Lennon 29 Although the autonomic dysfunction is mostly mild to moderate, and the frequency is similar in P-LES and NP-LES patients,Reference O’Neill, Murray and Newsom-Davis 28 the severity tends to be greater in older patients with cancer.Reference O’Suilleabhain, Low and Lennon 29 Adrenergic as well as cholinergic dysfunctions have been demonstrated in LES patients.Reference Khurana, Koski and Mayer 30 Cholinergic dysfunction affects predominantly the parasympathetic, but also the enteric and sympathetic autonomic nervous systems, either in isolation or in various combinations. A sluggish pupillary reflex is probably the most common sign of autonomic dysfunction (69%).Reference Clark, Newsom-Davis and Sanders 31 , Reference Wirtz, de Keizer, de Visser, Wintzen and Verschuuren 32 Xerostomia (73%) and erectile dysfunction (53% of men) are the most common complaints. Xerophtalmia, orthostatic hypotension, abnormal sweating, constipation and bladder dysfunction are less frequently reported. Ptosis, which may reflect combined defects in somatic and sympathetic neurotransmission, is found in up to 30% of cases.Reference Wirtz, Smallegange, Wintzen and Verschuuren 8 , Reference Waterman 33 The presence of subacute urinary retention or life-threatening abnormalities like severe orthostatic hypotension, gastroparesis with intestinal pseudo-obstruction or cardiac dysrhythmias all indicate the coexistence of a paraneoplastic autonomic neuropathy. Constitutional symptoms of anorexia and weight loss usually attributed to cancer may indicate an autoimmune gastrointestinal dysmotility. Paraneoplastic autonomic neuropathy is most commonly associated with anti-Hu (ANNA-1) antibodies or antibodies against the neuronal ganglionic acetylcholine receptor.Reference Benarroch 34 Early recognition of a concurrent autonomic neuropathy in LES is important, because paraneoplastic syndrome with autonomic neuropathy is associated with a worse prognosis.Reference Giometto, Grisold, Vitaliani, Graus, Honnorat and Bertolini 35
Physical Examination in LES
A cardinal feature of LES on examination is post-exercise potentiation of muscle strength, a short-lasting return toward the normal range after muscle contraction. Progressive augmentation of muscle strength is seen during the first few seconds of maximal voluntary contraction (MVC), but it is difficult to demonstrate on examination. Repeat testing over several seconds may be more effective, giving the examiner a sensation that with each successive effort the resistance needed to overcome the patient’s strength increases.Reference Kimura 36 In some LES patients with ptosis, a paradoxical improvement of ptosis with sustained upgaze has also been described,Reference Breen, Gutmann, Brick and Riggs 37 in contrast to the exercise-induced worsening of MG. However, continued prolonged voluntary effort does lead to fatigue, as seen in MG.
Tendon reflexes (TR) are typically depressed or absent in LES, even in muscles with relatively normal clinical strength. In contrast with LES, TR are characteristically normal in MG, whereas in myopathies TR are not affected until there is enough atrophic weakness.
The augmentation of TR, also elicited directly after a brief voluntary contraction (up to 10 s), is a unique and very useful clinical feature for diagnosing LES, although it is not very sensitive.Reference Odabasi, Demirci, Kim, Lee, Ryan and Claussen 38 TR may be preserved in mild LES.Reference Odabasi, Demirci, Kim, Lee, Ryan and Claussen 38 , Reference Zambelis, Foutsitzi, Giannakopoulou, Poulopoulou and Karandreas 39 Importantly, the presence of increased TR does not exclude LES, as it might be observed in LES patients with an associated myelopathy and/or encephalopathy. The physiological mechanisms in LES that explain the reason for the rare involvement of extraocular muscles, as well as the disparity between the severely affected TR and the relatively normal muscle strength, are nicely addressed by Meriggioli and colleagues.Reference Meriggioli 40
Electrodiagnostic Approach to LES
Electrodiagnostic studies are essential in diagnosing LES. As the syndrome’s electrical hallmark, routine motor nerve conduction stimulation typically elicits a very small low-amplitude compound muscle action potential (CMAP), frequently below 10% of the lower limit of normal, which is disproportional to the relatively preserved power. In contrast, motor conduction velocities and sensory studies are normal, unless there is an associated neuropathy. The CMAP amplitude represents the best parameter of severity in LES. As the patient’s condition improves, resting CMAP amplitude increases, decreasing the degree of facilitation.Reference Oh, Kim, Kuruoglu, Brooks and Claussen 41 - Reference Oh, Claussen, Hatanaka and Morgan 43
Low-rate (LRS) repetitive nerve stimulation (RNS) causes a decrementing CMAP response above 10% in LES; however, it is also found in MG patients. This response might be difficult to detect if initial CMAP amplitudes are severely reduced. Interestingly, in LES, RNS CMAPs typically continue to decrement from the first to the ninth intra-volley stimuli (“saddle-shape” decrement) with a smaller CMAP amplitude and a decrement greater than at rest (post-activation depression). This pattern has also been reported in muscle-specific kinase antibody-positive MG. In contrast, in seropositive acetylcholine receptor antibodies (AChRAb) MG, the decrement repairs after the fourth or fifth intra-volley stimulus (the classic “U-Shaped” decrement).Reference Sanders, Cao, Massey, Juel, Hobson-Webb and Guptill 44
Once LES is suspected, studies should look for facilitation, the most specific electrophysiological finding in LES. Potentiation, facilitation and increments, frequently used in the literature, are not interchangeable terms and may be confusing. Potentiation, as mentioned above, describes the incrementing mechanical muscle response elicited immediately during and after RNS (post-tetanic potentiation) or immediately at the end of an MVC (post-activation potentiation or post-exercise potentiation).Reference Kimura 45 In contrast, facilitation refers to an increase in an electrically measured response.Reference Kimura 46 Increments are calculated by comparing the highest CMAP amplitude or area with baseline CMAP following high-rate RNS (post-tetanic facilitation) or immediately after a brief MVC (post-activation facilitation, also called post-exercise facilitation). It is calculated as follows: Increment (%)=(Amplitude of Highest CMAP – Baseline CMAP/Amplitude of Baseline CMAP)×100.Reference Misra and Kalita 47
In LES, substantial increments in CMAP amplitude are observed following high-rate RNS or brief exercise for up to 10 seconds. A longer period of activation may lead to exhaustion and less facilitation. Care must be taken to ensure that the muscle has been completely rested for several minutes before testing, as even slight voluntary contraction may induce facilitation. Although post-exercise facilitation (PEF) is much less painful than post-tetanic facilitation, high-rate RNS is still useful in patients too weak to maximally contract, or when cooperation is suboptimal. Alternatively, a slower rate of stimulation also facilitates the response if combined with voluntary contraction.Reference LoMonaco, Milone, Padua and Tonali 48 Paired stimulation with an interstimulus interval of 5-10 ms causes the second response to increase rather than decrease, as expected in normal muscles.Reference Kimura 49
A CMAP increment >50% in any muscle suggests LES.Reference Brown and Johns 50 , Reference Juel and Sanders 51 However, care must be taken to ensure that any increase in CMAP is truly because of facilitation. Muscles stimulated repetitively at a high rate tend to discharge with increased synchrony without recruitment of additional muscle fibres. This phenomenon, called pseudofacilitation, characteristically increases amplitude, even by as much as 50%, decreasing CMAP duration and keeping the area of the waveform relatively constant.Reference Oh, Eslami, Nishihira, Sarala, Kuba and Elmore 52 Therefore, CMAP area measurements can be used to help distinguish pseudofacilitation from facilitation. Tim et al.Reference Tim and Sanders 53 addressed this issue: that the use of CMAP area instead of CMAP amplitude to measure the increment, in hand muscles, had no impact on the diagnostic performance of RNS.
The degree of increment needed to diagnose LES is controversial, as increments >100%, even as high as 300%, have also been reported in rare MG cases.Reference Singer, Smith, Ziegler and Festoff 54 , Reference Alboini, Damato, Iorio, Luigetti and Evoli 55 In MG, an incremental pattern is rarely found because the original CMAP amplitude is not or is only marginally reduced. Several cutoff values have been used for the diagnosis of LES. Oh et al.Reference Oh, Kurokawa, Claussen and Ryan 56 suggested that the diagnosis of LES be revised to a 60% increment criterion with a sensitivity of 97% for LES diagnosis and a specificity of 99% excluding MG. These authorsReference Oh, Hatanaka, Claussen and Sher 57 subsequently found that seropositive LES patients had a higher increment (meeting the 100% criterion), whereas those without VGCCAb only met the 60% criterion. Although no electrophysiological criteria will discriminate between all patients with MG and LES, it is accepted that when the increment is >400% in any muscle or >100% in most tested muscles, the patient almost certainly has LES.Reference Juel and Sanders 51 , Reference Tim and Sanders 53
LES patients show widely distributed electrophysiological abnormalities compared with MG, with variable electrical changes usually confined to clinically symptomatic muscles.Reference Maddison, Newsom-Davis and Mills 58 Nonetheless, the sensitivity of electrophysiological testing for LES diagnosis is substantially better in distal muscles compared with proximal muscles, and multiple muscles should be tested before concluding that the test is negative.Reference Tim, Massey and Sanders 11 RNS studies should include foot muscles, and studies of tibial and peroneal nerves may be crucial for confirming the diagnosis of LES in some cases.Reference Oh, Head, Fesenmeier and Claussen 59
Electrophysiological abnormalities may show various patterns, reflecting different degrees of blocking. Oh et al.Reference Oh 60 described three patterns in LES based on the RNS test, from the mildest in type 1 to the most severe in type 3. Type 2, considered the classical pattern, has been previously described (low CMAP amplitude, decrement >10% with LRS and increment >60%). Type 1 is characterized by normal or near-normal CMAP amplitudes, decremental responses at LRS and absence of CMAP facilitation. Type 3 consists of low CMAP amplitudes, a decrement >10% with LRS and post-tetanic facilitation with initial decremental responses. The last two patterns can be misinterpreted as MG patterns if prolonged stimulation is not performed. An overlap in this electrophysiological variability may be possible at different times during the course of the disease in the same patient or even at a given moment, within the same patient, depending on the severity of the disease or the muscles being tested.Reference Oh 60
Needle electromyography (EMG) may be useful in some scenarios. Short-duration polyphasic motor unit potentials (MUPs), observed in myopathies, are also described in LES.Reference Komatsu, Bokuda, Shimizu, Komori and Koide 61 Fibrillation potentials may also occur in severe cases.Reference Scola, Iwamoto, Ramos, Sanderson, Lopardo and Cantarelli 62 Findings of low-amplitude MUPs with marked waveform variability and an incrementing tendency in successive discharges in a patient with proximal lower limb weakness, who is being evaluated for possible myopathy, should arouse suspicion of LES.
Single-fibre EMG demonstrates abnormal jitter in virtually all patients with LES. Although this test is highly sensitive, it does not distinguish between pre- and post-synaptic localizations. Jitter is abnormal even in clinically strong muscles, and the degree of abnormality is frequently out of proportion to the severity of weakness, compared with MG.Reference Sanders and Stalberg 63 An improvement in abnormal jitter and blocking on increasing stimulation rate has been characteristically described in LES but has also been found in MG. Therefore, this pattern does not confirm the diagnosis of LES unless it is dramatic and is seen in most endplates.Reference Sanders 64 , Reference Trontelj and Stalberg 65 Jitter correlates well with clinical and electrophysiological severityReference Oh and Ohira 66 and can be used to monitor therapy.Reference Phillips 67
Respiratory Electrophysiological Evaluation
Respiratory muscle involvement in LES is probably under-recognized, and the available information is mostly confined to small retrospective case series and reports. Moreover, respiratory muscle weakness is usually documented based on clinical observation, arterial blood gas measurements and/or the assessment of respiratory mechanics.Reference Meriggioli 18 However, these evaluations are dependent on pulmonary factors, and it is therefore difficult to distinguish the status of the respiratory muscles from that of the lungs themselves. As alternatives, phrenic nerve conduction, RNS of the phrenic nerve and needle EMG of the diaphragm are valuable techniques that can be performed using standard EMG equipment. These methods are of clinical value in patients with undiagnosed dyspnea and those with known neuromuscular transmission defects.Reference Zifko, Nicolle, Grisold and Bolton 68 , Reference Zifko, Bolton and Nicolle 69 , Reference Colombo 71 Nicolle et al.Reference Nicolle, Stewart, Remtulla, Chen and Bolton 19 described two cases of LES who presented in the critical care unit with severe unexplained respiratory failure, reversible with treatment. The electrophysiological hallmarks—reduced or absent diaphragmatic CMAP (DCMAP) on phrenic nerve conduction, a decrement on phrenic LRS and a post-exercise facilitation of the diaphragm—led to the diagnosis of LES. Because of the influence of lung volume on the size and shape of DCMAPs, all trains of stimuli are given at the end of quiet expiration and patients are instructed to hold their breath during the train of stimuli. PEF is performed asking the patient to continually inspire through an incentive bedside spirometer for 10 seconds. Area, rather than amplitude, is the best measurement of neuromuscular transmission of the diaphragm because of the pseudofacilitation regularly observed in phrenic nerve RNS.Reference Zifko, Nicolle, Remtulla and Bolton 70 Diaphragmatic EMG is important to help rule out a disorder of central respiratory drive or denervation. Fibrillation potentials, positive sharp waves and a marked reduction of MUPs firing with each inspiration may be observed as a result of severe neuromuscular blocking. The degree of “functional” denervation can progressively decrease following treatment.Reference Nicolle, Stewart, Remtulla, Chen and Bolton 19
Electrophysiological Evaluation of Autonomous System
The autonomic tests should complement the battery of conventional electrophysiological techniques performed in the EMG laboratory. They are useful not only for diagnostic purposes but also to monitor and quantify those aspects of autonomic function that have an impact on outcome or evaluate treatment efficacy.Reference Colombo 71 Abnormal sympathetic and parasympathetic autonomic dysfunction has been noticed in LES. Establishing and maintaining the parasympathetic and sympathetic balance may slow the onset of cardiac autonomic neuropathy, thus minimizing morbidity and mortality risk.Reference Colombo 72
Autonomic dysfunction in LES can be demonstrated by quantitative sudomotor axon reflex testing, heart rate variability to a variety of stimuli, pupillary light responses, reflex tear production, orthostatic blood pressure changes or quantitative measurements of salivation and lacrimation.Reference O’Suilleabhain, Low and Lennon 29 - Reference Clark, Newsom-Davis and Sanders 31 , Reference Waterman 33 However, there are no large prospective clinical trials, and the information is based on several small case series. These tests require specialized equipment and trained personnel only available in a few neurophysiology laboratories.Reference Howard 73 , Reference Shahani, Day, Cros, Khalil and Kneebone 74 Alternatively, R–R interval variation (RRIV) at rest and during deep breathing, as well as sympathetic skin response (SSR), are simple, non-invasive tests of the parasympathetic and sympathetic functions, respectively, and are easily performed using standard EMG equipment. Although the validity of SSR remains questionable given the polysynaptic nature of the reflex and the interpretational limits,Reference Singer, Mauermann and Benarroch 75 its clinical utility in a wide variety of disorders has been well documented in numerous studies.Reference Claus and Schondorf 76 - Reference Gutrecht 79 High sensitivity and specificity in the evaluation of autonomic dysfunction have been reported when both tests are combined. Moreover, since some patients may be unable to perform deep breathing, measurements of RRIV at rest have still proven effective.Reference Shahani, Day, Cros, Khalil and Kneebone 74 Because RRIV and SSR evaluate different components of the autonomic nervous system, it is understandable that abnormalities do not always occur in parallel. The decreased R–R intervals on electrocardiogram (ECG) noted in some LES patients is a sign of a vagal function impairment. When it is combined with orthostatic hypotension, there may be an impairment of cardiovascular response. In such cases, clinicians should be cautious using drugs that produce myocardial depression or systemic vasodilatation.Reference Mason 80 The signs and symptoms of autonomic dysfunction improve in many patients with LES following the treatment of the underlying malignancy or with 3,4-diaminopyridine.Reference McEvoy, Windebank, Daube and Low 81
Although autonomic symptoms help to distinguish LES from MG, the mere presence of autonomic symptoms does not reliably do so. Clinicians should be aware of rare cases of MG with autoimmune autonomic ganglionopathy in patients with occult cancer.Reference Vernino, Cheshire and Lennon 82 , Reference Peltier, Black, Raj, Donofrio, Robertson and Biaggioni 83
Serological Approach for Diagnosis of LES
In clinical practice, P/Q-type VGCCAb have the highest sensitivity and specificity in LES, rather than the rarer N- or L-type VGCCAb. False-positive results are uncommon but may be attributable to repeated freezing and thawing of serum. VGCCAb have been anecdotally reported in other conditions, including MG, usually in low titres, and considered an epiphenomenon.Reference Lennon, Kryzer, Griesmann, O’Suilleabhain, Windebank and Woppmann 84 - Reference Leys, Lang, Johnston and Newsom-Davis 86 False-negative results, however, are possible after commencement of corticosteroid therapy.Reference Lennon 87
In a patient with seronegative AChRAb and oculobulbar muscle weakness, a positive test for VGCCAb may lead to the diagnosis of LES rather than MG.Reference Lennon 88 Similarly, the absence of autoantibodies combined with presynaptic electrophysiological abnormalities, rarely reported in some MG patients,Reference Alboini, Damato, Iorio, Luigetti and Evoli 55 may lead to overdiagnosis of LES. The distribution of clinical symptoms and a comprehensive electrophysiological assessment, including autonomic and stimulated single-fibre EMG (SFEMG) testing, can help to distinguish between both conditions. Serological retesting could also be useful, as seronegative LES patients may seroconvert as the disease progresses.Reference Juel and Sanders 89 One helpful observation is that in LES the electrophysiological abnormalities are extensively distributed and are usually more severe than the clinical findings would suggest, whereas the opposite is often true in MG. In addition, a diagnosis of LES is generally accepted if the CMAP increment exceeds 100% in most of the muscles tested or 400% in any muscle.Reference Juel and Sanders 51 Notwithstanding the above, continued observation and retesting will be necessary in some patients before achieving a correct diagnosis.Reference Harper and Lennon 90 It has practical consequences in light of the potential for unnecessary treatments or delayed detection of an underlying malignancy.
The sensitivity and specificity for the VGCCAb assay are affected by the source of the antigen and the specific laboratory performing the measurements. The differences reported in the literature about the incidence of P/Q-type VGCCAb in LES patients may also be explained by different methods in defining the cutoff.Reference Motomura, Lang, Johnston, Palace, Vincent and Newsom-Davis 91 P/Q-type VGCCAb are usually detected at ≥100 pmol/L in 95% of non-immunosuppressed LES patients. Values >20 pmol/L are considered positive by some authors, as fewer than 3% of healthy participants in their studies had these antibodies.Reference Lennon, Kryzer, Griesmann, O’Suilleabhain, Windebank and Woppmann 84 , Reference Lennon 87
In conclusion, a positive P/Q-type VGCCAb result is useful for distinguishing LES from MG, but results must be interpreted in the context of the clinical and electrophysiological findings in each individual patient. Additionally, not all patients have seropositive VGCCAb, and results are often severely delayed in those who do. However, the neurophysiologic evaluation is the most definitive diagnostic test available and is highly distinctive.Reference Meriggioli 92
Differential Diagnoses of LES
The differential diagnosis of LES includes MG, congenital myasthenic syndromes and defects of neuromuscular transmission induced by drugs or toxins. A congenital myasthenic syndrome is severe at birth. The diagnosis of botulism is usually made on clinical grounds and confirmed by electrodiagnostic studies and demonstration of the toxin in the stool. The combination of proximal weakness and reduced TR, seen in LES, might suggest an inflammatory neuropathy with minimal sensory involvement or myopathy. Proximal muscle weakness is the most common presentation of a wide range of myopathies. In patients with “myopathic” presentation, normal creatine kinase and negative AChRAb, the presence of very low CMAP amplitudes and autonomic symptoms points toward LES. Patients with critical illness neuropathy and/or myopathy may have low CMAP amplitudes without sensory abnormalities. Because of the available treatments with neuromuscular-blocking effects, a high-rate RNS is mandatory to rule out LES. MG is the main differential diagnosis of LES, and Table 1 outlines the major differences between LES and MG. They can usually be distinguished by differences in clinical manifestations as well as autoimmune, serological and electrodiagnostic tests. As positive edrophonium test and favourable response to acetylcholinesterase inhibitors are characteristically reported in MG, they can help distinguish both conditions. However, a definitive response to acetylcholinesterase inhibitors is also noted in some LES patients.Reference Henriksson, Nilsson, Rosen and Schiller 93 - Reference Dell’Osso, Ayyar, Daroff and Abel 95
Finally, cases of an overlap or combination of LES and MG have been reported in the literature; see Oh et al. for a review.Reference Oh 96
Illustrative Cases
Case 1
In April of 1985, a previously healthy 7-year-old boy was hospitalized at the Hospital Universitario La Fe (Valencia) following a two-month history of weakness with difficulties in climbing stairs. The neurological examination revealed proximal leg weakness and a waddling gait with pronounced lumbar lordosis. TR were diminished in the upper extremities and were absent in the lower limbs. Cerebrospinal fluid, brain magnetic resonance imaging and nerve conduction studies were normal. An EMG suggested myopathy, but a muscle biopsy was normal. The weakness improved without treatment over several days, and he was discharged without additional work-up. RNS and an autoantibody panel were not performed. Follow-up examinations were conducted at an outpatient clinic, and the symptoms spontaneously resolved within two months. No conclusive diagnosis was reached.
In January of 2003, at the age of 25 years, the patient was referred to our clinic under the suspicion of MG. He complained of a one-month history of generalized fatigue and leg-predominant weakness following an exacerbation of psoriatic skin lesions. He again complained of difficulty in rising from a chair and in climbing stairs. Other symptoms included postural dizziness, dry eyes and mouth, and exercise-induced aching of his pelvic-girdle muscles. A neurological examination showed bilateral and symmetrical ptosis, without ocular motor disturbances or oropharyngeal weakness. A paradoxical lid elevation was observed immediately after 10 seconds of sustained upgaze (Fig. 1A). Sluggish pupillary reflex and generalized hyporeflexia were noted on examination. The strength examination was relatively normal without fatigue. No atrophy or fasciculations were noted. Motor nerve conduction studies showed low CMAP amplitudes in three muscles: the abductor digiti minimi (ADM), right extensor digitorum brevis (EDB) and left orbicularis oculi (O. Oculi). In the left EDB and right abductor digiti quinti pedis muscles, the CMAP amplitudes were near the lower limit of normal. Motor nerve conduction velocities and sensory nerve conduction studies were normal. Bilateral phrenic conduction showed normal DCMAP amplitudes and latencies. Needle EMG on quadriceps showed polyphasic MUPs with short durations, low amplitudes and marked instability. These results, along with the clinical symptoms and signs on examination, out of proportion to the patient’s complaints, suggested the diagnosis of LES. LRS showed a “saddle-shape” decrement in all muscles. The PEF criterion was met in all muscles, except in both hemi-diaphragms and O. Oculi muscle. The highest increment (>400%) was noted in the right EDB muscle (Table 2 and Fig. 2). RNS at 50 Hz was required to demonstrate a significant increment in left O. Oculi muscle (Table 2 and Fig. 1B). Paired stimulation (8-ms interstimulus interval) of right common peroneal nerve elicited an increased second response in the EDB muscle. SFEMG on extensor digitorum communis and O. Oculi revealed a markedly abnormal jitter and blocking with 3 Hz of axonal stimulation that dramatically improved at high frequencies (30-40 Hz) without blocking. Parasympathetic and sympathetic autonomic function were assessed by RRIV and SSR, respectively. Decreased R–R variation was noted at rest (6.8; normal: 18.9±7.2) and during forced deep breathing (11.5; normal: 31.0±9.3). SSRs were elicited in lower and upper limbs.
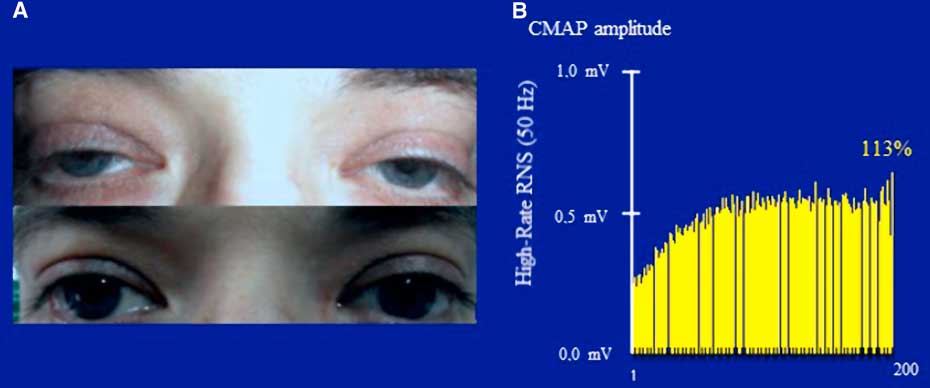
Figure 1 Case 1. Panel A: A clinical hallmark of LES is the improvement of strength within seconds of a brief maximal voluntary contraction. The image shows an improvement of ptosis after a brief upgaze. Note the symmetry of ptosis in sharp contrast to MG, where it is usually asymmetrical, frequently varying from one side to the other. Panel B: Prolonged high-rate stimulation of the left facial nerve. Notice the initial low-amplitude CMAP (recorded from the orbicularis oculi) and the significant post-tetanic facilitation after a 5-s train of stimulation with 50 Hz.
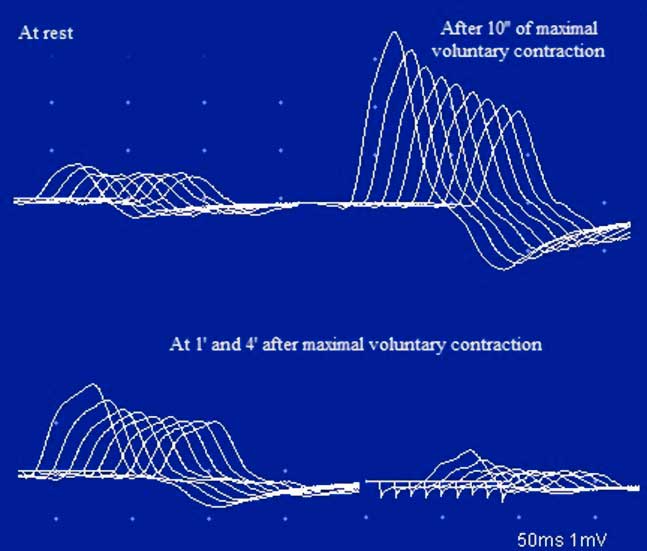
Figure 2 Case 1. An RNS study of the ulnar nerve at 3 Hz. Notice the low CMAP amplitude of the ADM, with a decrement and post-tetanic facilitation after 10 s of maximal voluntary contraction. There are lower-amplitude responses with decrements at 1 min and even 4 min after the maximal voluntary contraction. The CMAP amplitude decreases progressively from the first to the ninth stimulus, with a smaller CMAP amplitude and a decrement greater than at rest (post-activation exhaustion). This pattern is characteristic of LES.
Table 2 Electrophysiological Studies Conducted Before and After Treatment
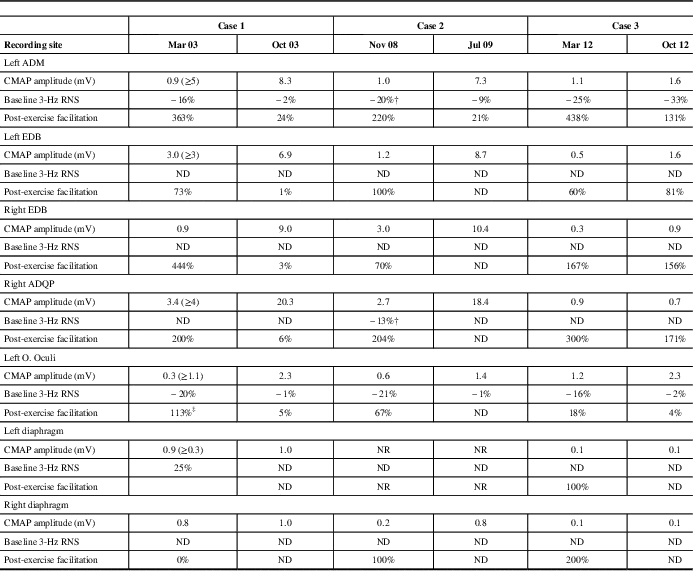
‡ High-rate stimulation (50 Hz); † “U-shaped” train envelope (3 Hz); RNS=repetitive nerve stimulation; ADM=abductor digiti minimi; EDB=extensor digitorum brevis; ADQP=abductor digiti quinti pedis; O. Oculi=orbicularis oculi; ND=not done; NR=non-response; CMAP=compound muscle action potential. Normal values in parentheses.
Serological assays for anti-smooth muscle antibodies, anti-striated muscle antibodies and AChRAb (<0.1; cutoff value: >0.2 nmol/L) and P/Q-type VGCCAb (<5; cutoff value: ≥25 pmol/L) factors were negative. Thyroid hormone levels were normal. Thorax/abdomen computed tomography (CT) and whole-body fluorodeoxyglucose-positron emission tomography (FDG-PET) were unremarkable.
The patient was diagnosed with LES and received pyridostigmine (180 mg daily) and deflazacort (30 mg daily), which resulted in marked clinical improvement. Resting CMAP amplitudes recovered, without decremental response to LRS or facilitation, and RRIV testing became normal over 7 months (Table 2 and Fig. 3). No stimulated SFEMG was performed. The patient felt almost entirely clinically recovered but still complained of some difficulty in climbing stairs. Following the neurologist’s instructions, the patient began corticosteroid withdrawal while maintaining the pyridostigmine. A gradual improvement was felt and, in February of 2004, the patient did not report any complaints. A gradual pyridostigmine discontinuation was initiated. A further clinical and electrophysiological assessment in January of 2005 was normal, confirming the spontaneous remission. Serological AChRAb (0.04 nmol/L) and VGCCAb (<5 pmol/L) titres were normal. To date, the patient remains asymptomatic and cancer-free.
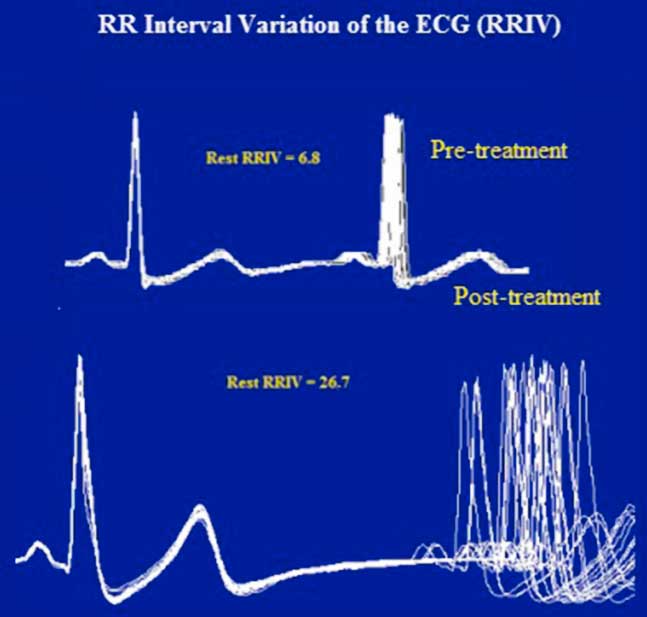
Figure 3 Case 1. Resting heart rate variability. Like the RNS tests, the abnormal R–R interval variability of the ECG was also resolved after treatment. The R–R interval is displayed by superimposing 20 sweeps triggered by the QRS complex of the electrocardiogram. The jitter (top) in the second potential is expressed as a percentage of the mean interpotential interval (bottom) as follows: RRIV=a/b×100.
Case 2
In September of 2008, a 50-year-old man presented to the emergency department following a syncopal episode. He was a 50-pack/year cigarette smoker who had experienced dyspnea, asthenia, a weight loss of 18 pounds and vague muscle stiffness with aching for 8 weeks. A chest radiograph showed a left hilar enlargement with main stem bronchus occlusion and lower lobe atelectasis. A CT scan and bronchoscopy with biopsy confirmed SCLC at a tumour–node–metastasis clinical stage of T4N3M1. A palliative chemotherapy regimen (cis-platinum and VP-16) was started in October of 2008. During the following weeks, the patient complained of difficulty in walking. The gait was characterized by wide-based steps with unsteadiness. Motor and coordination examinations were normal, but TR were unobtainable. A diagnosis of chemotherapy-related neuropathy versus paraneoplastic sensory neuropathy was initially considered.
In November of 2008, he was referred to our department with the suspicion of myopathy. For a week, he had experienced fluctuating lower limb weakness, crawling sensations in his muscles and difficulties getting out of a chair. The neurological examination found a waddling gait. It seemed to improve initially on exercise but then gradually declined with sustained exercise, with the patient complaining of “fatigue”. Motor examination revealed symmetrical proximal weakness (4/5 on arm abduction at the shoulder and hip flexors) without fatigue. The sensory examination was normal. In striking contrast with the absence of significant muscle atrophy or weakness, TR were absent. These findings, together with their being out of proportion to the patient’s complaints, raised the suspicion of LES. The clinical hallmark of LES, the augmentation of TR elicited immediately after a brief voluntary contraction, was subsequently confirmed in the upper and lower limbs (see the Video). Then, the patient was interrogated about the presence of autonomic symptoms, revealing a dry mouth with an unpleasant “metallic taste”, dry eyes, erectile dysfunction and constipation. The patient also reported self-limiting episodes of dizziness and blurred vision, without complete loss of consciousness. Such episodes had occurred frequently prior to hospital admission. Orthostatic hypotension testing showed that the blood pressure was 143/84 mmHg supine and 78/59 mmHg in the standing position, and caused similar symptoms.
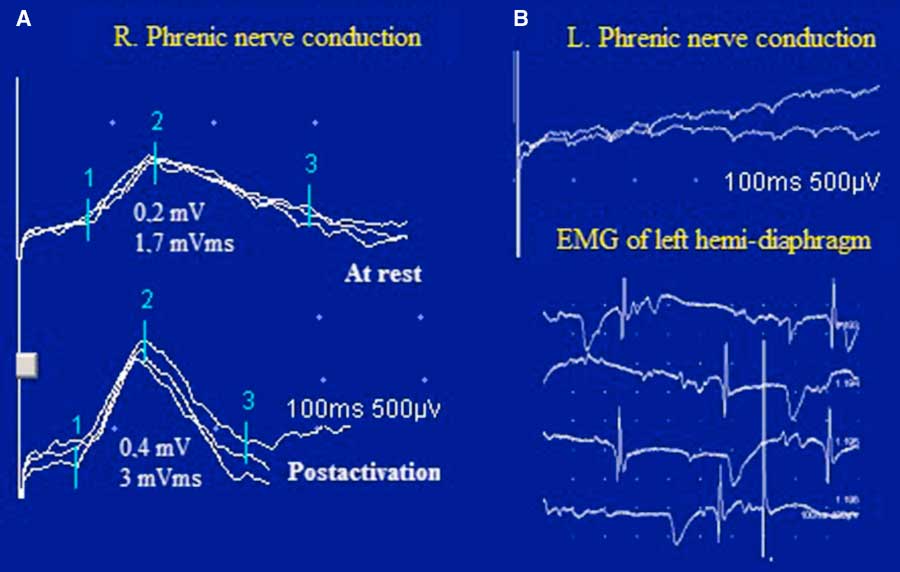
Figure 4 Case 2. Bilateral phrenic nerve conduction and diaphragmatic needle EMG. In panel A, notice the low amplitude and the area of the right diaphragmatic CMAP with PEF after inspiration through an incentive spirometer for 10 s. In panel B, there was no response in the left hemi-diaphragm to phrenic nerve stimulation and the diaphragmatic needle EMG showed profuse fibrillation potentials and positive sharp waves.
The electrophysiological studies confirmed the classical pattern expected in LES: diffuse very low CMAP amplitudes with normal motor conduction velocities, decrement with LRS and diffuse PEF ranging between 67 and 220%. A “U-shaped” decrement at LRS, typically reported in AChRAb positive MG, was noted in two muscles (Table 2 and Fig. 5A). High-rate RNS at 20 Hz in the left ADM muscle resulted in nonsignificant post-tetanic facilitation (36%) with initial decremental responses, mimicking an MG pattern (Fig. 5B). However, immediately after brief maximum voluntary contraction of the left ADM muscle, CMAP amplitude increased >200% (Table 2).
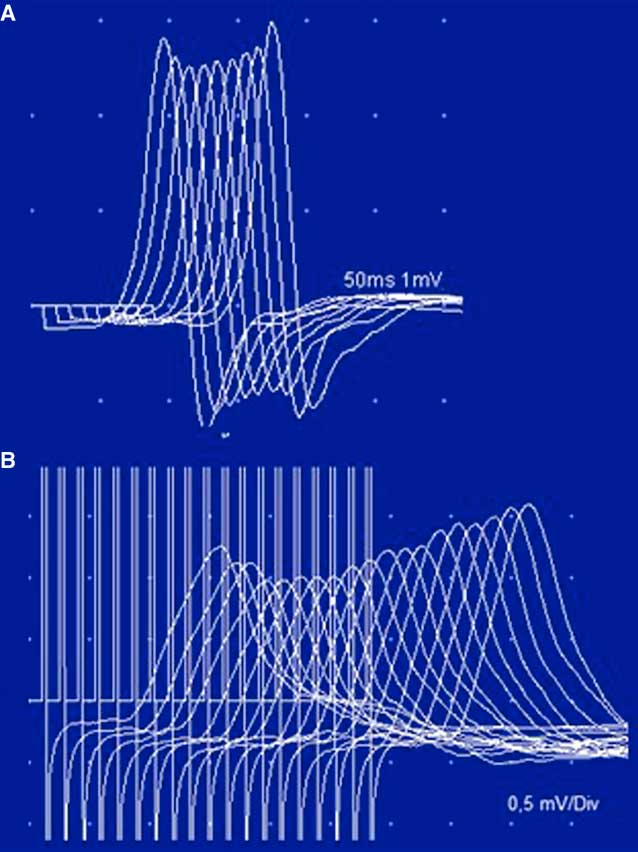
Figure 5 Case 2. Unusual stimulation patterns. Panel A: The low-rate stimulation of the tibial nerve shows a “U-shaped” decrement in the abductor digiti quinti pedis, typically described in AChRAb-positive MG. Panel B: High-rate stimulation (20 Hz) in the left ulnar nerve. The low CMAP amplitude and the initial decrement (14.3%) were not followed by a significant incremental response (36%). This pattern has been reported in a few cases of severe MG. Note that a 220% incremental response was obtained after brief exercise (see also Table 2).
Bilateral phrenic nerve conduction revealed absent left phrenic nerve response and reduced DCMAP amplitude (0.2 mV; normal value: >0.3 mV) and negative area (1.7 mVms; normal value: >2 mVms) on the right, with normal latency (7.6 ms; normal value: >8.2 ms). A 100% increment in DCMAP amplitude and a 76% increment in the negative area were observed immediately after inspiration through an incentive spirometer (DATOSPIR 120, SibelMed, Barcelona, Spain) for 10 seconds. Needle EMG of the left hemi-diaphragm demonstrated diffuse fibrillations and positive sharp waves and no MUPs, consistent with functional denervation, severe neuromuscular blocking or axonal degeneration from malignant invasion of the phrenic nerve (Figure 4).
A paraneoplastic antibody panel was slightly positive for AChRAb (0.3 nmol/L) but showed high titres of P/Q-type voltage-gated calcium channel antibodies (343 pmol/l). LES was diagnosed, and the patient was prescribed 3,4-diaminopyridine (20 mg, four times a day), which elicited significant clinical improvement. A new neurophysiologic assessment in July of 2009 confirmed normal resting CMAP amplitudes in upper and lower extremity muscles and left O. Oculi. LRS of the ADM muscle elicited a normal CMAP amplitude, a borderline decrement of 9% and no significant incremental response after brief exercise (Table 2). The neurophysiologic assessment of phrenic nerve and diaphragm showed persisting findings on the left side, although normal results were observed for the right phrenic nerve. These findings suggested a persistent phrenic nerve injury in which a neuromuscular transmission defect was effectively treated in the right nerve only. Unfortunately, six months later, the cancer progressed rapidly and the patient died.
Case 3
In March of 2012, a 73-year-old woman presented to our electrodiagnostic laboratory with the suspicion of myopathy. She reported a 17-year history of lower back pain, exercise-induced muscle aching, vague leg paresthesia and difficulty walking. Previous evaluations in other institutions suggested different diagnoses, such as lumbar spinal stenosis, sensory polyneuropathy, myopathy and even “psychogenic weakness”. Proximal weakness with difficulty climbing stairs, dry mouth, blurred vision because of dry eyes, episodes of syncope and constipation had developed progressively over the years. During the two years preceding referral to our clinic, these symptoms got progressively worse with the additional appearance of a waddling gait, difficulty chewing, neck extensor weakness and dyspnea on exertion. The results of pulmonary function tests and chest radiographs were normal. Her medication included artificial tears for chronically dry eyes, celecoxib and chondroitin sulphate for leg pain and domperidone for the episodes of dizziness upon standing.
A neurological examination showed mild proximal weakness without fatigue. The arm reflexes were 1+. TR were obtainable in the lower limbs after a brief MVC. Electrodiagnostic studies of multiple muscles were consistent with LES. CMAP amplitude in the O. Oculi muscle was borderline. A progressive decremental pattern was obtained in response to LRS. Diagnostic facilitation could not be demonstrated (Table 2). The tests also showed diaphragmatic and parasympathetic autonomic dysfunction. The results of the following laboratory tests were normal: thyroid hormones, Sry-type high-mobility group box (SOX) and assays for anti-phospholipid, anti-smooth muscle, anti-striated muscle and anti-acetylcholine receptor antibodies. The P/Q-type VGCCAb assays were positive (264.7 pmol/l). Chest radiography, whole-body CT scan and FDG-PET scans were normal. The patient was prescribed 3,4-diaminopyridine (10 mg, four times a day). Difficulty chewing, muscle aching, cardiac R–R abnormalities and most autonomic symptoms were completely resolved following treatment. Dyspnea and neck and limb weakness were significantly relieved with minimal resolution of the electrophysiological abnormalities. To date, the patient is cancer-free.
Conclusions
These cases illustrate how, although LES is considered a rare disorder, many cases may go unrecognized because clinical, neurophysiologic and/or serological features overlap with other more common disorders or predominance of non-muscle symptoms.
In the first patient, a rare case of transient seronegative NP-LES associated with psoriasis, the early ocular involvement initially suggested MG. The symmetrical pattern of ptosis with improvement after sustained upgaze, sluggish pupillary reflexes and the post-exercise potentiation of TR pointed toward LES. Unlike in MG, the electrophysiological abnormalities were widely distributed in multiple muscles without apparent involvement. Facilitation greater than 400% in any muscle or greater than 100% in most tested muscles, as noted in this patient, is almost certainly diagnostic of LES. Interestingly, the peroneal nerve was critical in confirming the diagnosis of LES and demonstrated that multiple distal muscles should be tested before reaching a conclusive diagnosis. The satisfactory clinical response to cholinesterase inhibitors documented, in line with previous reports, that it is not necessarily diagnostic of MG and can be observed in LES.
In case 2, a patient with seropositive LES initiated following cancer diagnosis and “myopathic” presentation, the findings of abnormal TR and very low CMAP amplitudes in contrast to nearly normal muscle strength, post-exercise TR potentiation and striking autonomic symptoms pointed toward LES. The presence of low titres of AChRAb showed how the serological profile should be interpreted in the context of the individual patient’s clinical and electrophysiological findings. The respiratory electrophysiological investigation was invaluable in confirming a left phrenic nerve axonopathy and a severe but reversible presynaptic blockade of neuromuscular transmission of the right hemi-diaphragm. As observed in case 2, misleading MG-like RNS patterns can be noted in a patient with LES, depending on the muscle tested. Both cases 1 and 2 showed that facilitation assessment should be included in the diagnostic algorithm of unexplained weakness, even if the CMAP amplitudes are borderline or normal (Table 2). As seen in these cases, the diagnostic accuracy of post-tetanic versus post-exercise facilitation varied depending on the muscle tested (Table 1, Fig. 5).
In case 3, the predominance of non-muscle symptoms led to unnecessary diagnostic work-ups and treatments and delayed a correct diagnosis for years. This case illustrated how marked electrophysiological abnormalities can be noted in a paucisymptomatic or even asymptomatic LES patient. RRIV testing proved to be a reliable test of parasympathetic autonomic function for diagnostic purposes and also to evaluate treatment efficacy in cases 1 and 3. Finally, spontaneous remission (case 1), although rare compared with MG, is possible in LES.
Ethics Committee Approval
Informed written consent was obtained for all patients. The study was exempt from ethics committee approval, as it was comprised entirely of retrospective and observational data analysis of previously treated patients. Moreover, current clinical best practice guidelines were adhered to in each case, and no additional risks were imposed on the patients.
Funding
This case study received no specific grant from any funding agency in the public, commercial or not-for-profit sectors.
Disclosures
Miguel Angel Merino-Ramírez and Charles Bolton hereby declare that they have no conflicts of interest to disclose.
Statement of Authorship
Dr. Merino-Ramírez was involved in the acquisition of data and is the first author of the manuscript. Drs. Merino-Ramírez and Bolton conceived of and designed the study. Both authors had full access to all of the data in the study, and they take responsibility for the integrity and accuracy of the data and their interpretation. Both authors were involved in preparing the manuscript. They both have critically reviewed all aspects of the submitted manuscript and given their full approval.
Supplementary material
To view supplementary material for this article, please visit http://dx.doi.org/10.1017/cjn.2016.268








