



Introduction
Frontotemporal dementia (FTD) is the term used for a group of clinical syndromes that differ from Alzheimer’s disease (AD) by presenting at a younger age (mean, 60 years), progressing more rapidly and more often having an underlying genetic cause. Reference Pottier, Ravenscroft, Sanchez-Contreras and Rademakers1,Reference Woollacott and Rohrer2 Although much less common than AD, FTD is an important neurodegenerative disease that accounts for 10%–15% of all dementia and is the second most common type before age 65. Reference Onyike and Diehl-Schmid3 With an estimated prevalence of 15/100,000, there are currently ∼50,000 individuals suffering from FTD in North America, resulting in significant socio-economic costs. Reference Galvin, Howard, Denny, Dickinson and Tatton4 FTD is a multifaceted clinical syndrome, characterized by progressive deterioration in behaviour, personality and/or language, with relative preservation of memory, early in the disease. Reference Woollacott and Rohrer2 Clinical subtypes include the behavioural variant (bvFTD) and two forms of primary progressive aphasia (PPA), progressive non-fluent aphasia (PNFA or nfvPPA) and semantic dementia (SD or svPPA). Reference Gorno-Tempini, Hillis and Weintraub5,Reference Rascovsky, Hodges and Knopman6 In addition, there is growing recognition that FTD shows significant clinical, pathological and genetic overlap with a number of movement disorder syndromes; particularly corticobasal syndrome (CBS), progressive supranuclear palsy (PSP) and motor neuron disease (MND). Reference Lomen-Hoerth, Anderson and Miller7,Reference Padovani, Agosti, Premi, Bellelli and Borroni8
Although there is currently no effective treatment for FTD, major recent advances in our knowledge of the molecular genetic and pathological basis Reference Pottier, Ravenscroft, Sanchez-Contreras and Rademakers1,Reference Mackenzie and Neumann9 have provided exciting opportunities to develop targeted, disease-modifying therapies. Reference Ljubenkov and Boxer10,Reference Tsai and Boxer11 However, testing and implementing this type of precision medicine requires accurate, early diagnosis, which is especially complicated in FTD due to the high degree of clinical and pathological heterogeneity, factors that emphasize the urgent need for refined diagnostic criteria and the development of useful biomarkers. The aim of this review is to describe the current state of FTD diagnosis and its challenges, and briefly summarize the remarkable molecular advances that have been made, in order to provide useful context for understanding the rationale behind recent biomarker and therapeutic strategies.
Terminology
For most of the 20th century, the term ‘Pick’s disease’ (PiD) was broadly used for cases of non-AD dementia with lobar atrophy, regardless of the underlying pathology. Reference Constantinidis, Richard and Tissot12 By the 1980’s, researchers in select centres had begun to focus specifically on these cases and accrue sizable cohorts, which allowed for the publication of a number of position papers that attempted to clarify the nosology and better define the clinical and pathological criteria. Reference McKhann, Albert and Grossman13–15 Although most recommended the term ‘FTD’ to refer to the clinical syndrome, and ‘frontotemporal lobar degeneration’ (FTLD) for the underlying pathologies, Reference Mackenzie, Neumann and Bigio16 others have used ‘FTLD’ more generically. The diagnosis of PiD is now usually reserved for relatively rare cases of clinical FTD with pathologically confirmed Pick bodies.
Clinical FTD Subtypes
The clinical manifestations of FTD reflect the preferential neurodegeneration of the frontal and temporal cerebral lobes. In an effort to improve diagnostic accuracy, new clinical diagnostic criteria were developed in the last decade for each of the FTD subtypes (Table 1 and Table 2), Reference Gorno-Tempini, Hillis and Weintraub5,Reference Rascovsky, Hodges and Knopman6 as well as the overlapping motor syndromes of PSP, CBS and FTD-MND. Reference Armstrong, Litvan and Lang17–Reference Strong, Abrahams and Goldstein19
Table 1: Criteria for bvFTD (modified from Rascovsky et al.) Reference Rascovsky, Hodges and Knopman6
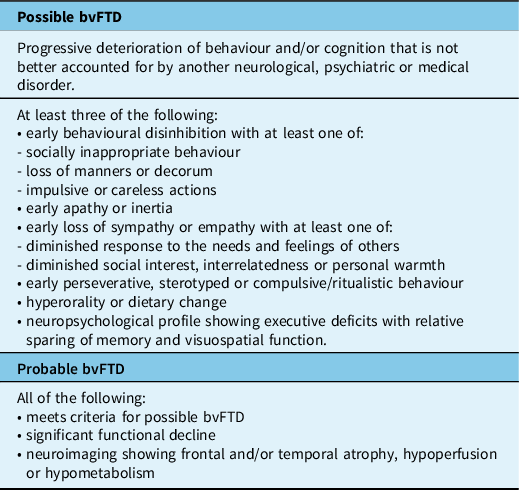
Table 2: Criteria for PPA subtypes (modified from Gorno-Tempini et al.) Reference Gorno-Tempini, Hillis and Weintraub5
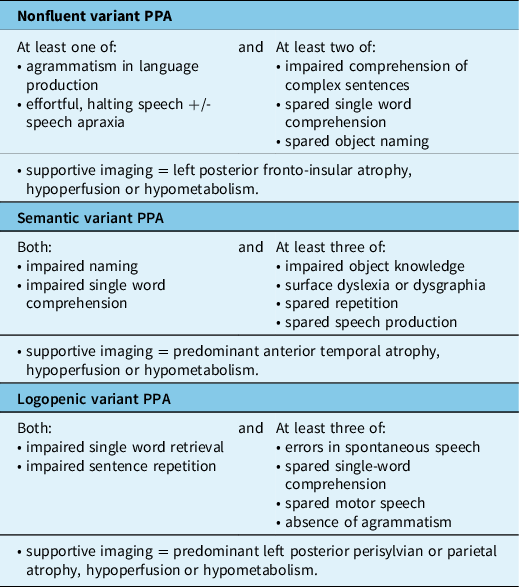
Behavioural Variant FTD (bvFTD)
BvFTD is the most common clinical subtype, accounting for approximately half of all FTD cases. Reference Coyle-Gilchrist, Dick and Patterson20,Reference Johnson, Diehl and Mendez21 The mean age of onset is approximately 58 years and the average survival is 8 years; however, there is wide variation, with earlier onset and more rapid course in those with a genetic cause, and much shorter survival in those with associated amyotrophic lateral sclerosis (ALS). Reference Caswell, McMillan and Xie22,Reference Lomen-Hoerth23 BvFTD is characterized by progressive and persistent changes in personality and behaviour with decline in social skills and executive dysfunction. Reference Woollacott and Rohrer2,Reference Rascovsky, Hodges and Knopman6 Typical early symptoms include apathy, disinhibition, repetitive and/or compulsive behaviours, change in eating habits and decreased empathy. Deficits in executive function result in difficulties planning and higher level thinking; whereas, language, visuospatial skills and memory are usually preserved, early in the disease. Although not specifically included in the current diagnostic criteria by Rascovsky et al., Reference Rascovsky, Hodges and Knopman6 impaired social cognition (‘theory of mind’) and emotion recognition are also common features. Reference Torralva, Gleichgerrcht, Torres Ardila, Roca and Manes24 The most recent consensus criteria provide three levels of diagnostic certainty with ‘possible’ bvFTD based upon the presence of core symptoms alone, ‘probable’ including supportive neuroimaging findings (see above) and ‘definite’ bvFTD for cases with proven pathology or causal mutation (Table 1). Reference Rascovsky, Hodges and Knopman6 These new criteria have improved diagnostic sensitivity and specificity (possible bvFTD, 85%–95% and 82%, respectively; probable bvFTD, 75%–85% and 95%, respectively). Reference Balasa, Gelpi and Martín25,Reference Harris, Gall and Thompson26 BvFTD is the most common presentation of all the major genetic causes of FTD and may be associated with all of the common molecular pathologies (i.e. FTLD-tau, FTLD-TDP and FTLD-FET) (Table 3). Reference Mackenzie and Neumann9
Table 3: Molecular classification of FTLD with genetic and clinical correlations (modified from Hodges FTD) Reference Neumann, Kovacs, Mackenzie and Dickerson40
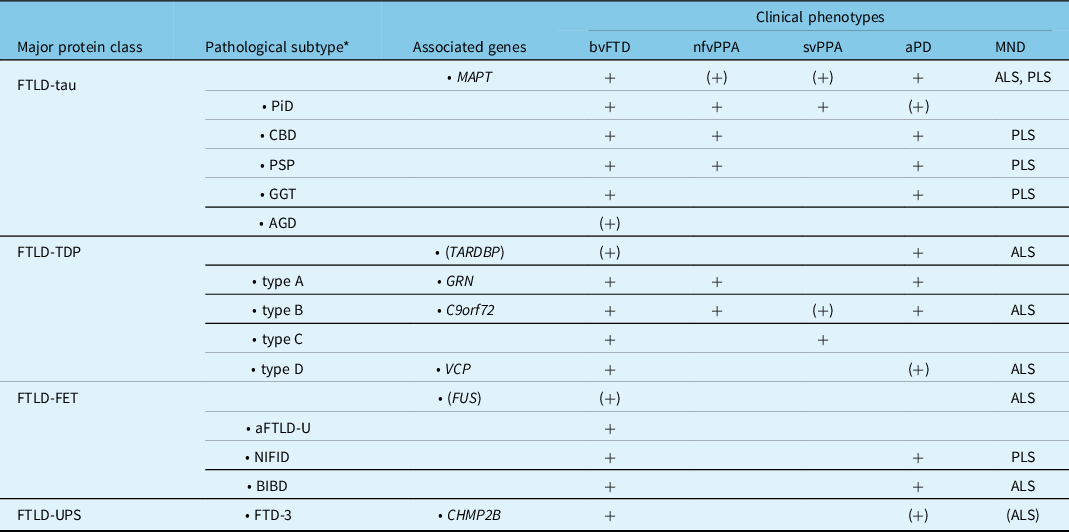
aFTLD-U, atypical frontotemporal lobar degeneration with ubiquitinated inclusions; AGD, argyrophilic grain disease; ALS, amyotrophic lateral sclerosis; aPD, atypical parkinsonism; BIBD, basophilic inclusion body disease; bvFTD, behavioral variant FTD; C9orf72, chromosome 9 open reading frame 72 gene; CBD, corticobasal degeneration; CHMP2B, charged multivescicular body protein 2B gene; FTD, frontotemporal dementia; FTD-3, FTD linked to chromosome 3; FTLD, frontotemporal lobar degeneration; FUS, fused in sarcoma gene; GGT, globular glial tauopathy; GRN, progranulin gene; MAPT, microtubule associated protein tau gene; MND, motor neuron disease; nfvPPA, non-fluent primary progressive aphasia; NIFID, neuronal intermediate filament inclusion disease; PiD, Pick’s disease; PLS, primary lateral sclerosis; PSP, progressive supranuclear palsy; svPPA, semantic variant PPA; TARDBP, transactive response DNA binding protein gene; TDP, TDP-43; UPS, ubiquitin proteasome system; VCP, valosin containing protein gene.
(+)Rare cause or unusual phenotype.
* Indicates the characteristic pattern of pathology, not the clinical syndrome.
Primary Progressive Aphasia (PPA)
A diagnosis of PPA is appropriate when progressive language decline and speech difficulties are the most prominent presenting features and remain predominant during the first few years of disease. Reference Gorno-Tempini, Hillis and Weintraub5,Reference Mesulam27,Reference Mesulam28 The non-fluent and semantic variants (nfvPPA and svPPA, respectively) are usually associated with FTLD pathology and, therefore, considered as part of the FTD clinical spectrum; whereas, logopenic variant PPA (lvPPA) is usually caused by AD-type pathology (Table 2). Although most cases of PPA can be classified as one of these three common variants, some cases present with a broader spectrum of language dysfunction and have been referred to as mixed PPA. Reference Mesulam, Wieneke, Rogalski, Cobia, Thompson and Weintraub29
Non-fluent Variant PPA (nfvPPA)
Non-fluent (or agrammatic) PPA is characterized by expressive language deficits in which speech is slow, effortful, telegraphic and is often accompanied by agrammatism, phonemic paraphasias and speech apraxia. Reference Gorno-Tempini, Hillis and Weintraub5 There is sparing of single word comprehension, but an impaired understanding of complex sentences. Personal and interpersonal conduct, behaviour and insight are preserved early in the disease. NfvPPA accounts for 25% of FTD and has a slightly later mean age at onset (63 years) and an intermediate rate of progression (9 years mean survival). Reference Coyle-Gilchrist, Dick and Patterson20,Reference Johnson, Diehl and Mendez21 It may be associated with either underlying FTLD-tau or FTLD-TDP and is a relatively common presentation in families with GRN or C9orf72 mutations (Table 3). Reference Mackenzie and Neumann9
Semantic Variant PPA (svPPA)
SvPPA presents with anomia, impaired single word comprehension and surface dyslexia, due to loss of semantic memory, but sparing of speech production (i.e. fluency). Reference Gorno-Tempini, Hillis and Weintraub5 Initial word-finding difficulties often involve low frequency or specialized terms, but progressively includes words that are more common. SvPPA accounts for approximately 20% of all FTD cases and has a mean onset of 60 years and a relatively slow progression (11 years mean survival). Reference Coyle-Gilchrist, Dick and Patterson20,Reference Johnson, Diehl and Mendez21 In typical cases, atrophy of the left anterior temporal lobe underlies the progressive loss of meaning for words, objects and emotions. Less commonly, cases with predominant right anterior temporal lobe atrophy present with behavioural changes similar to bvFTD, memory impairment, prosopagnosia and only mild, late-onset language deficits. Reference Ulugut Erkoyun, Groot and Heilbron30 These ‘right semantic dementia’ or ‘right temporal variant FTD’ cases also have more frequent depression, somatic complaints and mental slowness. SvPPA is usually sporadic and is almost always associated with FTLD-TDP type C pathology (Table 3). Reference Mackenzie and Neumann9
Logopenic Variant PPA (lvPPA)
LvPPA, characterized by word-finding difficulties, anomia and impaired sentence repetition, is most often an atypical presentation of AD and not considered part of the FTD spectrum. Reference Gorno-Tempini, Hillis and Weintraub5,Reference Rohrer, Rossor and Warren31
Molecular Neuropathology and Genetics of FTD
The neuropathology that underlies clinical FTD is heterogeneous. Reference Mackenzie and Neumann9 Degeneration of the frontal and temporal cerebral lobes is a relatively consistent feature and FTLD is used as the general term for those pathological conditions that commonly present as clinical FTD. Reference Mackenzie, Neumann and Bigio16 Until quite recently, our knowledge of the molecular basis of FTD was limited to a subset of cases in which the pathology is characterized by abnormal intracellular accumulation of the microtubule-associated protein tau (FTLD-tau), which includes cases with PiD, PSP and CBD pathology, and families with autosomal dominantly inherited FTD and parkinsonism caused by mutations in the tau gene (MAPT) (Figure 1). Reference Ghetti, Wszolek, Boeve, Spina, Goedert, Dickson and Weller32 However, this situation changed dramatically, beginning in 2006, when mutations in another gene on chromosome 17 (GRN), that encodes the progranulin protein (PGRN), were also found to cause familial FTD. Reference Baker, Mackenzie and Pickering-Brown33,Reference Cruts, Gijselinck and van der Zee34 That same year, the pathological protein in most tau-negative cases was identified as being the transactive response DNA binding protein Mr 43 kD (TDP-43). Reference Neumann, Sampathu and Kwong35 It was subsequently found that the small number of cases without either tau or TDP-43 pathology had cellular inclusions composed of fused in sarcoma (FUS) and the other FET proteins. Reference Neumann, Rademakers, Roeber, Baker, Kretzschmar and Mackenzie36,Reference Neumann, Bentmann and Dormann37 Most recently, abnormal expansion of a GGGGCC hexanucleotide repeat in a non-coding region of the chromosome 9 open reading frame 72 gene (C9orf72) was identified as being the most common genetic cause of both FTD and ALS. Reference DeJesus-Hernandez, Mackenzie and Boeve38,Reference Renton, Majounie and Waite39 These and other recent discoveries now make it possible to assign virtually all cases of clinical FTD to one of three pathological protein classes (FTLD-TDP, 50%; FTLD-tau, 45%; FTLD-FET, 5%) Reference Mackenzie and Neumann9 and to determine the underlying genetic cause in most families with inherited FTD (Figure 2). Reference Pottier, Ravenscroft, Sanchez-Contreras and Rademakers1
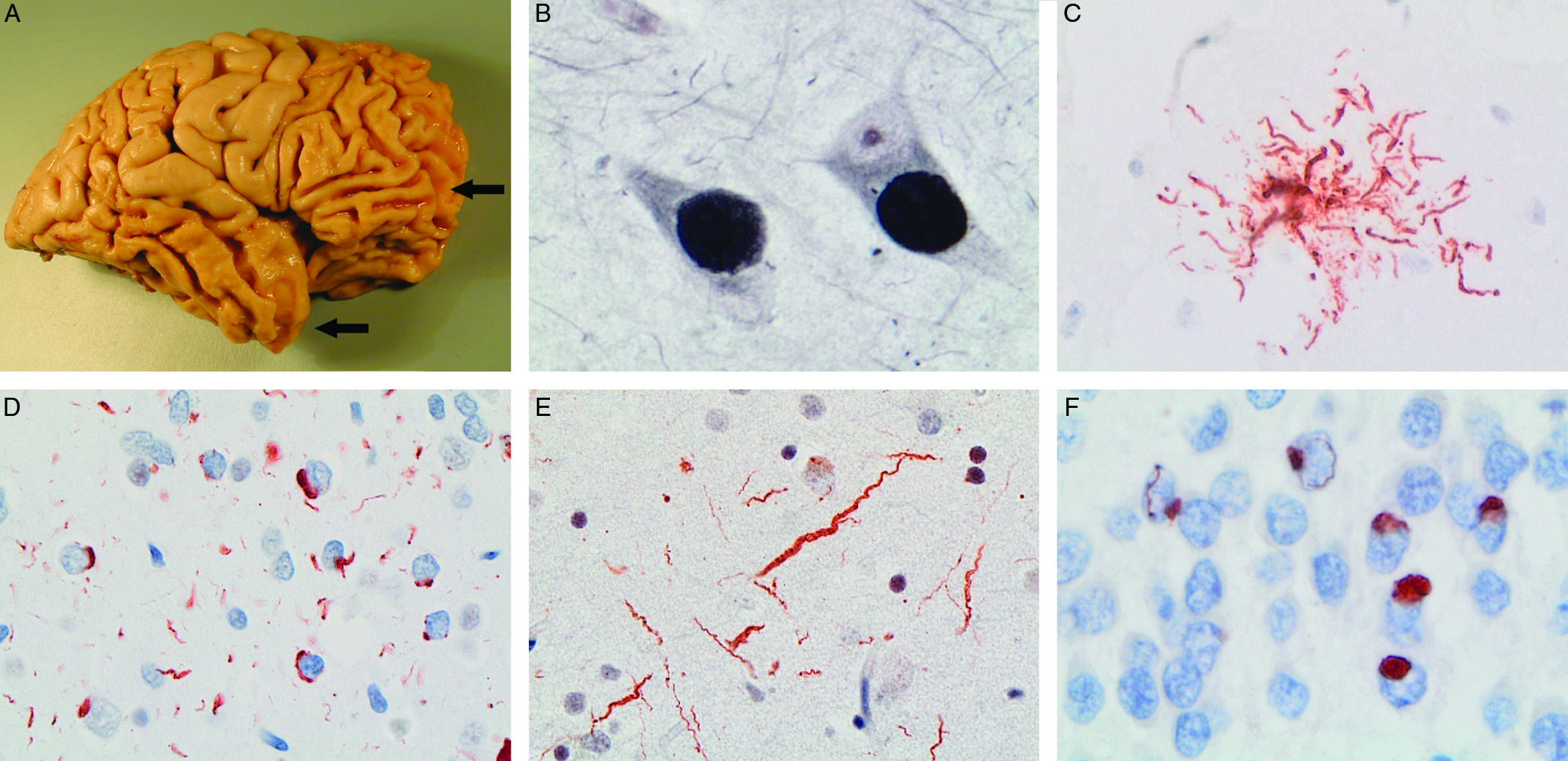
Figure 1: The neuropathology underlying clinical FTD is variable. There is usually preferential atrophy of the frontal and/or temporal cerebral lobes (arrows) (A) and the term frontotemporal lobar degeneration (FTLD) is used for pathological conditions that commonly present as clinical FTD. Examples of FTLD with cellular inclusions composed of tau protein (FTLD-tau) include classical Pick’s disease (PiD) with neuronal Pick bodies (B), progressive supranuclear palsy with tufted astrocytes (C) and corticobasal degeneration (not shown). Different patterns of TDP-43-immunoreactive pathology include FTLD-TDP type A, characterized by neuronal cytoplasmic inclusions and short neurites, found in most cases of familial FTD caused by GRN mutations (D) and FTLD-TDP type C with long, tortuous neurites, present in most cases of semantic variant PPA (e). A small proportion of cases have cellular inclusions composed of FUS and other FET proteins (FTLD-FET) (F). (A) Gross photo of postmortem brain of patient with behavioural variant FTD and classical PiD, (B) Pick bodies in pyramidal neurons of hippocampus stained with Bielschowsky silver method, (C) tufted astrocyte labelled with tau immunohistochemistry (IHC), (C, E) TDP-43 IHC on sections of frontal cortex, (F) FUS IHC on section of hippocampal granule cell layer.

Figure 2: Molecular basis of FTD. FTLD-TDP pathology is present in approximately half of clinical FTD cases, including those caused by mutations in the chromosome 9 open reading frame 72 (C9orf72), granulin (GRN) and a number of other genes. FTLD-tau pathology is slightly less common (∼45%) and is found in all familial cases with tau gene (MAPT) mutations. A small proportion of sporadic FTD cases have pathological inclusions composed of FUS and other FET proteins (FTLD-FET).
Associated Motor Syndromes
Over the past few decades, there has been increasing recognition that FTD shows significant overlap with a number of movement disorder syndromes at a clinical, genetic and pathological level (Table 3). Patients who present with FTD may subsequently develop extrapyramidal features, often with just bradykinesia and rigidity, Reference Padovani, Agosti, Premi, Bellelli and Borroni8 but sometimes with more complex syndromes, such as PSP or CBS. Reference Kertesz, McMonagle and Jesso41 Similarly, corticobasal degeneration (CBD) and PSP pathology can both be associated with aphasia (usually nfvPPA) Reference Kertesz, McMonagle, Blair, Davidson and Munoz42,Reference Peterson, Patterson and Rowe43 or significant frontal lobe dysfunction, Reference Burrell, Hodges and Rowe44 and these cognitive features are now included in the clinical diagnostic criteria for CBS and PSP. Reference Armstrong, Litvan and Lang17,Reference Höglinger, Respondek and Stamelou18 Pathogenic mutations in MAPT, which are always associated with tau-based pathology, may manifest clinically as either FTD or atypical parkinsonism. Reference Ghetti, Wszolek, Boeve, Spina, Goedert, Dickson and Weller32 Less often, mutations in genes that result in FTLD-TDP (e.g. C9orf72, GRN) may have extrapyramidal features that are usually mild and late-onset, but may occasionally be the presenting feature. Reference Anor, Xi, Zhang, Moreno, Sato, Rogaeva and Tartaglia45–Reference Tremolizzo, Bertola, Casati, Piperno, Ferrarese and Appollonio48
A similar strong relationship exists between FTD and MND. Approximately 10% of patients who present with ALS go on to fulfill diagnostic criteria for dementia, often with features of FTD (more often bvFTD than PPA), and up to 50% of ALS patients have evidence of more subtle frontal lobe dysfunction on imaging or neuropsychological testing. Reference Strong, Abrahams and Goldstein19,Reference Lomen-Hoerth23,Reference Lomen-Hoerth, Murphy, Langmore, Kramer, Olney and Miller49 Conversely, up to half of patients with FTD are found to have signs of pyramidal dysfunction, with some eventually fulfilling criteria for either ALS or primary lateral sclerosis (PLS). Reference Lomen-Hoerth, Anderson and Miller7 TDP-43 is the pathological protein in more than 95% of clinical ALS, and FTLD-TDP is the most common pathological substrate for FTD. Reference Mackenzie and Rademakers50 Many (but not all) mutations that are associated with TDP-43 pathology can present as either FTD or MND, or both; specifically, C9orf72 and TBK1 mutations commonly present as ALS or PLS, but GRN mutations do not. Reference DeJesus-Hernandez, Mackenzie and Boeve38,Reference Freischmidt, Wieland and Richter51,Reference Gass, Cannon and Mackenzie52 As a result, CBS/CBD and PSP are sometimes included within the broader spectrum of FTD, and FTD and ALS are often considered to represent a clinical spectrum of disease with overlapping pathogenesis.
Challenges in Diagnosis and Prognosis
Accurate, early diagnosis is crucial to providing patients and their families with useful information about their disease, avoiding unnecessary or inappropriate treatment, and selecting patients for relevant clinical trials. However, even with application of the revised clinical diagnostic criteria, distinguishing FTD from other common neurodegenerative and psychiatric conditions remains challenging. Reference Balasa, Gelpi and Martín25,Reference Harris, Gall and Thompson26 Prominent FTD-like behavioural abnormalities are not uncommon in AD, particularly in young-onset cases (i.e. frontal or behavioural variant AD). The clinical subtypes of PPA may be difficult to distinguish from one another and do not consistently predict underlying FTLD versus AD pathology. Reference Chare, Hodges and Leyton53 Early memory impairment, which was previously considered to be an exclusionary criteria for FTD, Reference Neary, Snowden and Gustafson14 is increasingly recognized as a feature in some patients with FTD and may be sufficient to meet AD diagnostic criteria. Reference Fernández-Matarrubia, Matías-Guiu and Cabrera-Martín54,Reference Hornberger and Piguet55 It is also fairly common for young FTD patients with prominent personality changes and behavioural abnormalities to receive an incorrect initial diagnosis of a primary psychiatric disorder, such as schizophrenia, bipolar disorder or major depression, with FTD only becoming recognized as the disease progresses. Reference Ducharme, Dols and Laforce56,Reference Woolley, Khan, Murthy, Miller and Rankin57
Additional challenges result from the clinical and pathological heterogeneity within FTD. Although the initial presentation may be a relatively pure FTD subtype, additional behavioural, language, psychiatric and/or motor features often emerge and accumulate as the disease progresses, resulting in a convergence of FTD phenotypes. Even within families, where affected members share the same causal mutation, there may be significant variation in the age at onset, presenting features and disease course. Reference Benussi, Padovani and Borroni58 This variation in the natural history make it difficult to determine prognosis and to establish outcome measures and endpoints for clinical trials.
Finally, although some FTD clinical syndromes have strong pathological correlations (e.g. PSP with tau pathology, svPPA and FTD-ALS with TDP-43 proteinopathy), and each of the common FTD-causing mutations has a consistent underlying pathology, it is not possible to accurately predict the FTLD molecular subtype in patients with the most common FTD presentations of sporadic bvFTD and nfvPPA (Table 3). These challenges underscore the urgent need for biomarkers that support early diagnosis, help monitor disease progression and accurately predict the underlying pathology.
Biomarkers
Neuroimaging
The patterns of volume loss seen with structural neuroimaging (T1-weighted MRI) help to distinguish FTD from other neurodegenerative conditions and to differentiate the various FTD clinical subtypes (Figure 3). Frontotemporal atrophy with relative sparing of the hippocampus and medial temporal lobes has been reported to distinguish FTD from AD with a sensitivity of 55%–94% and specificity of 81%–97%. Reference Boccardi, Laakso and Bresciani59–Reference Likeman, Anderson and Stevens61 Cases with bvFTD show particular involvement of bilateral frontal lobes, anterior insula and anterior/mid cingulate, with anterior temporal, striatum and thalamus also frequently involved. Reference Pan, Song and Yang62 However, there is significant heterogeneity among individual cases and cluster analysis suggests that there may be several neuroanatomical subtypes of bvFTD, showing either focal or diffuse, frontal versus temporal-dominance patterns. Reference Whitwell, Przybelski and Weigand63 Patients with svPPA have a characteristic pattern of asymmetric atrophy of anterior and inferior temporal regions with the left temporal pole often affected early. Reference Gorno-Tempini, Dronkers and Rankin64,Reference Rohrer, Warren and Modat65 As disease progresses, orbitofrontal, inferior frontal, insula and anterior cingulate may also become affected and the contralateral hemisphere may be involved. In nfvPPA, there is left perisylvian atrophy affecting frontal operculum, dorsolateral prefrontal, superior temporal and insula. Reference Gorno-Tempini, Dronkers and Rankin64 Progression is associated with spread to ipsilateral anterior frontal, lateral temporal and anterior parietal regions and the right prefrontal and temporal lobes. Reference Rohrer, Warren and Modat65,Reference Rogalski, Cobia, Harrison, Wieneke, Weintraub and Mesulam66
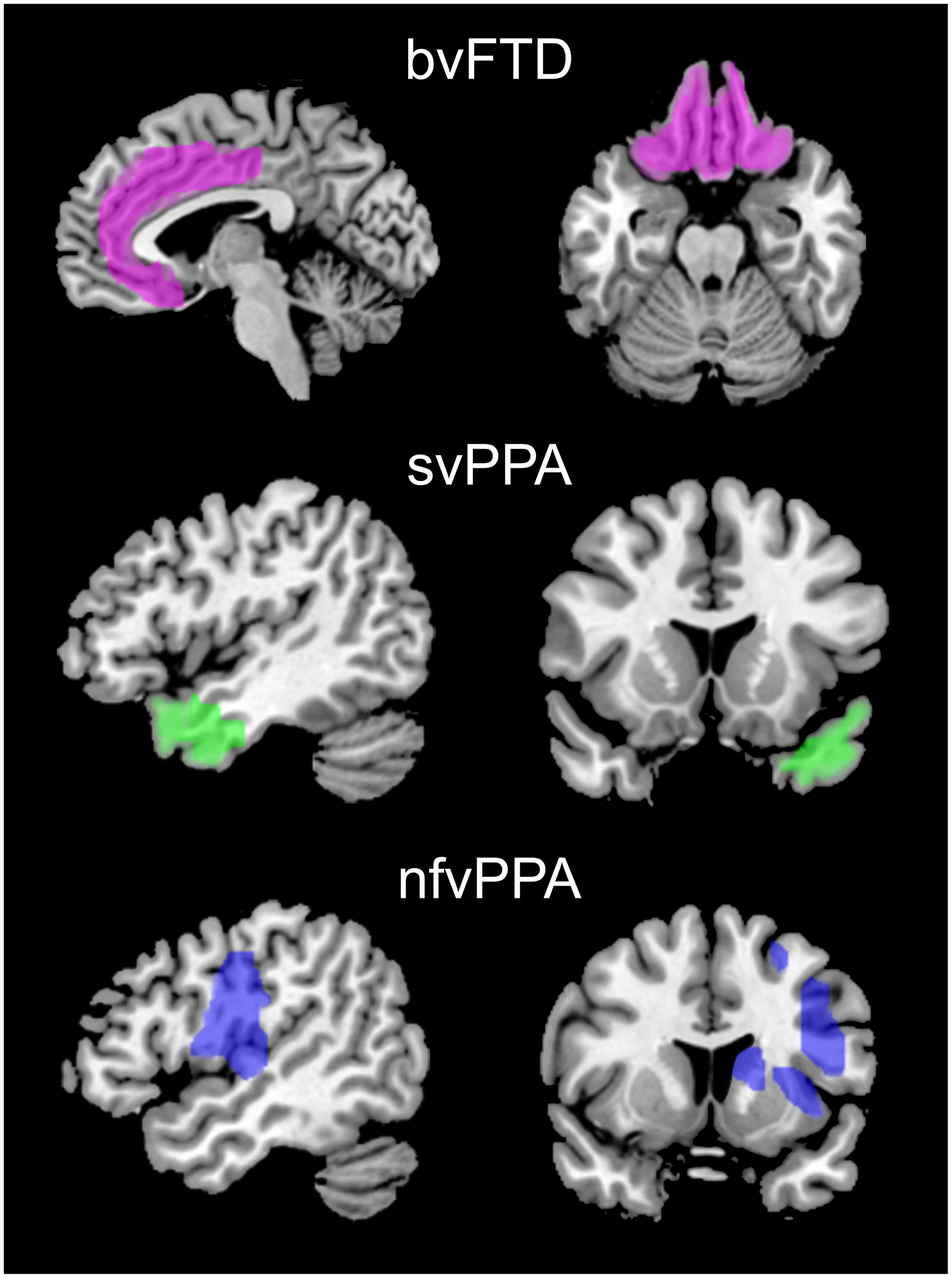
Figure 3: Characteristic patterns of atrophy in FTD subtypes seen with structural neuroimaging. Patients with bvFTD show bilateral atrophy of mesial and orbital frontal regions (pink). In svPPA, atrophy is lateralized (left>right) and targets the anterior temporal lobes and temporal poles (green). nfvPPA is characterized by atrophy of the left insula, frontal operculum, dorsolateral prefrontal and superior temporal lobes (blue).
In addition to clinical correlations, each of the main genetic and pathological subtypes of FTD displays characteristic patterns of volume loss. Reference Whitwell, Weigand and Boeve67 Longitudinal studies of inherited forms of FTD have provided important insights into the natural history of the disease and have demonstrated that neuroimaging changes can be detected many years (even decades) prior to the onset of symptoms, with each of the major genetic groups (MAPT, GRN and C9orf72) showing different rates and sequential patterns of involvement. Reference Rohrer, Nicholas and Cash68
Imaging modalities that reflect brain metabolism (e.g. FDG-PET) and perfusion (e.g. SPECT) tend to show regional patterns of change that correlate with and may precede atrophy in FTD patients. In particular, FDG-PET distinguishes FTD from AD with high sensitivity and specificity (97% and 86%, respectively) Reference Foster, Heidebrink and Clark69,Reference Rabinovici, Rosen and Alkalay70 and may be more sensitive than MRI for detecting early changes in certain genetic FTD subtypes. Reference Popuri, Beg and Lee71 More novel imaging techniques, such as diffusion tensor imaging (DTI), resting-state functional MRI and arterial spin labelling are proving useful in research investigating changes in functional connectivity within brain networks. Reference Gordon, Rohrer and Fox72
Particularly promising is the development of PET-based imaging tracers that bind to and demonstrate the anatomical distribution of specific pathological protein aggregates. PET imaging of the amyloid-beta protein that forms AD-type senile plaques differentiates AD from FTD with high sensitivity and specificity, although the proportion of positive FTD cases may increase with age due to the presence of coexisting AD and FTLD pathology. Reference Rabinovici, Rosen and Alkalay70,Reference Villemagne, Ong and Mulligan73 Newly developed radioligands that recognize pathological forms of tau protein are also proving useful for diagnosing AD. Reference Ossenkoppele, Rabinovici and Smith74 Although tau PET has the potential to distinguish between the major pathological subtypes of FTD (FTLD-tau vs. FTLD-TDP), preliminary studies suggest that the currently available ligands (e.g.18F-AV-1451/Flortaucipir F18) are most sensitive for the paired helical filament tau that forms neurofibrillary tangles in AD and less so for the common FTLD-tau pathologies such as PSP and CBD. Reference Tsai, Bejanin and Lesman-Segev75 This is perhaps not unexpected in light of recent cryo-electron microscopy findings that the pathological tau filaments that aggregate in each of the major tauopathy conditions are composed of different tau amino acid residues and have differently folded structures. Reference Shi, Zhang and Yang76 However, there also appear to be issues of specificity since off-target binding in structures such as the basal ganglia and substantia nigra is common and positive signals have been reported in some patients with clinical or genetic forms of FTD that are commonly associated with TDP-43 pathology. Reference Tsai, Bejanin and Lesman-Segev75,Reference Leuzy, Chiotis and Lemoine77 More recently, early studies of a number of second-generation tau tracers (e.g. PI-2620) have shown some promising results. Reference Cassinelli Petersen, Roytman, Chiang, Li, Gordon and Franceschi78 In addition to being sensitive for AD tau pathology, these have less off-target binding and some have demonstrated increased uptake in disease-relevant brain regions in patients suspected of having 4R tauopathies (e.g. PSP and CBS). Although the urgent need to develop an imaging biomarker specific for TDP-43 pathology is also well-recognized, there has been little progress to date. Finally, a number of studies have shown that combining multiple neuroimaging modalities (MRI and FDG-PET or DTI) can improve classification accuracy when distinguishing FTD from other neurodegenerative disorders and psychiatric disease. Reference Tahmasian, Shao and Meng79,Reference Vijverberg, Wattjes and Dols80
Biofluid Biomarkers
Most neurochemical biomarkers for neurodegenerative disease are measured in CSF, although the development of ultrasensitive assays (e.g. single molecule array, Simoa) is making it possible to detect some brain-derived proteins in serum and plasma.
The most important differential diagnosis for FTD is usually AD. The neuropathology of AD is characterized by the accumulation of Aβ42 protein in senile plaques and hyperphosphorylated tau in neurofibrillary tangles and the measurement of these proteins in CSF is now commonly used in the diagnostic workup of AD, where Aβ42 is reduced while total tau (t-tau) and phosphorylated tau (p-tau181) are increased. Reference Jack, Albert and Knopman81 In FTD, Aβ42 and p-tau181 are typically normal while t-tau may be normal or elevated Reference van Harten, Kester and Visser82 and a number of studies have shown that an elevated ratio of t-tau:Aβ42 or p-tau181:Aβ42 (i.e. AD CSF profile) is particularly useful for differentiating AD from FTD, especially in younger patients (sensitivity 70%–89%, specificity 70%–94%). Reference Rivero-Santana, Ferreira and Perestelo-Pérez83
Biomarkers that accurately predict the specific biochemical type of pathology in individuals with FTD are currently lacking. It is perhaps surprising that CSF tau levels have not proven to be more useful in differentiating FTD clinical subtypes or in identifying those with underlying FTLD-tau. However, most studies have failed to demonstrate elevated p-tau181 levels in FTD patients with predicted or proven tau pathology, Reference Irwin, Lleó and Xie84,Reference Rosso, van Herpen and Pijnenburg85 and although the ratio of p-tau181:t-tau has been found to be higher in FTLD-tau compared to FTLD-TDP, Reference Borroni, Benussi and Archetti86,Reference Hu, Watts and Grossman87 this appears to be due to elevated t-tau in some FTLD-TDP patients with more severe neuronal loss. Studies measuring novel tau fragments that may be more disease-specific are ongoing but have been inconclusive, thus far. Reference Foiani, Cicognola and Ermann88 Developing useful bioassays of TDP-43 is also proving to be a challenge. A number of studies have used various techniques to evaluate full-length and pathological forms of TDP-43 in the CSF and blood, and although some have shown elevated levels in patients with FTD or ALS, the results have been inconsistent, often with significant overlap among disease and control groups. Reference Feneberg, Gray, Ansorge, Talbot and Turner89
These limitations highlight the need for more sensitive and specific assays to distinguish the various FTLD-related proteinopathies. One recently developed method that is showing promise is ‘real-time quaking-induced conversion’ (RT-QuIC). Reference Atarashi, Sano, Satoh and Nishida90 There is growing evidence that the proteins that aggregate and characterize most neurodegenerative conditions, including the FTLDs, each have an abnormal physical conformation that is unique to that pathological entity. Reference Hock and Polymenidou91 Disease progression is thought to result from ‘prion-like’ propagation as pathological protein molecules act as seeds that direct the mis-folding of native isoforms in a template-dependent manner. RT-QuIC detects the various disease-specific protein isoforms based on their seeding activity in an artificial assay. Reference Atarashi, Sano, Satoh and Nishida90 Early studies indicate that RT-QuIC may be able to detect the presence of pathological forms of TDP-43 and tau proteins in patient CSF and may even distinguish among different tau pathological subtypes (e.g. 3-repeat tau of PiD vs. 4-repeat tau of PSP and CBD). Reference Saijo, Ghetti and Zanusso92–Reference Scialò, Tran and Salzano94
There is also currently great interest in a number of proteins that are non-specific indicators of neurodegeneration and/or neuroinflammation as potential biomarkers of neurodegenerative disease activity. Of these, neurofilament light chain (NfL) is emerging as one of the most useful. Reference Ashton, Janelidze and Al Khleifat95 When axons are damaged, neurofilament proteins are released into the extracellular space and then move into the CSF and blood, where neurofilament levels are thought to reflect the overall severity of neurodegeneration, but not the specific cause. Reference Petzold96 NfL is particularly abundant, easy to measure and there is strong correlation between levels in CSF and blood. NfL levels are elevated in all clinical FTD subtypes, particularly in those who also have ALS. Reference Landqvist Waldö, Frizell Santillo and Passant97,Reference Rohrer, Woollacott and Dick98 Although NfL levels are significantly higher in FTD than other common causes of dementia, such as AD and Lewy body disease, there is significant overlap, and the discriminatory power is best when NfL is combined with other neuroimaging or biofluid biomarkers. Reference Zhao, Xin, Meng, He and Hu99 NfL is particularly useful for distinguishing FTD from non-neurodegenerative conditions (e.g. psychiatric disease) where levels are usually normal. Reference Al Shweiki, Steinacker and Oeckl100 NfL is also a good predictor of the rate of disease progression in FTD and may be useful for monitoring the efficacy of disease-modifying therapies. Reference Rohrer, Woollacott and Dick98
Finally, there are a few biomarkers that are proving to be useful in specific genetic subtypes of FTD. Heterozygous mutations in GRN are one of the most common causes of familial FTD, with all causal mutations resulting in a 50% reduction in the levels of PGRN protein. Reference Gass, Cannon and Mackenzie52 Immunoassays can detect reduced levels of PGRN in CSF and blood, and accurately predict the presence of a pathogenic GRN mutation, even in pre-symptomatic mutation carriers. Reference Finch, Baker and Crook101,Reference Galimberti, Fumagalli and Fenoglio102 In addition to diagnostic utility, monitoring PGRN levels may be useful in clinical trials of PGRN modulating therapies for this FTD patient population (see below). The most common genetic cause of both FTD and ALS is abnormal expansion of a hexanucleotide repeat sequence in the C9orf72 gene. Reference DeJesus-Hernandez, Mackenzie and Boeve38,Reference Renton, Majounie and Waite39 Even though the repeat is located in a non-coding region of the gene, the massive pathogenic expansions trigger unconventional translation of the repeat sequence that generates a set of five dipeptide repeat (DPR) proteins that accumulate in brain tissue and may be neurotoxic. Reference Ash, Bieniek and Gendron103,Reference Mori, Weng and Arzberger104 Elevated levels of one of these DPR proteins (poly-glycine-proline, GP) can be quantitatively detected in CSF by ELISA in mutation carriers. Reference Gendron, Chew and Stankowski105 Although the sensitivity and specificity of poly-GP assays are insufficient for genetic diagnosis and levels do not correlate with disease activity, measurements may be useful for monitoring the effect of treatments designed to silence the unconventional translation (e.g. antisense oligonucleotides). Reference Gendron, Chew and Stankowski105
Current and Future Treatment
Treatment for FTD currently revolves around the education of patients and caregivers, behavioural interventions and management strategies for bvFTD, and speech therapy for PPA. Reference Ljubenkov and Boxer10,Reference Tsai and Boxer11 Antidepressant, antipsychotic and anti-epileptic medications are often used to manage certain behavioural features in FTD patients, despite the absence of good clinical trial support. There is little biological evidence or clinical trial data to support the use of currently available AD medications, such as cholinesterase inhibitors and NMDA antagonists, in FTD. Novel symptomatic treatments that are currently being investigated include the use of the hormone oxytocin for treating social apathy Reference Finger, MacKinley and Blair106 and transcranial electrical and magnetic stimulation. Reference Benussi, Dell’Era and Cosseddu107
Recent advances in our knowledge of the molecular basis and pathomechanistic aspects of FTD have prompted a shift in focus towards potential disease-modifying therapies. A number of strategies for reducing the toxic effects of abnormal tau aggregation have been the subject of recent or ongoing clinical trials for AD and conditions associated with FTLD-tau. Reference Jadhav, Avila and Schöll108 These tau-focussed therapies include efforts to (i) promote the clearance of pathological tau aggregates with immunotherapy, either passive immunization with anti-tau antibodies or active immunization with vaccines containing tau peptides, (ii) inhibit pathogenic posttranslational modifications and aggregation of tau, (iii) reduce the expression of tau using antisense oligonucleotides (ASO) and (iv) compensate for the loss of normal tau function with molecules that promote microtubule stability.
Other recent clinical trials are focussing on preventing or modifying the disease course of specific genetic subtypes of FTD. Strategies to therapeutically elevate PGRN levels in patients with pathogenic GRN mutations include the use of small molecules to upregulate transcription of the wild-type GRN allele, Reference Cenik, Sephton and Dewey109 gene therapy approaches to replace the null-allele using viral vectors, Reference Hinderer, Miller and Dyer110 and suppressing PGRN uptake and degradation with antibodies that block the sortilin receptor. Reference Lee, Almeida and Prudencio111 Finally, intrathecal ASO therapy targeting the expanded C9orf72 RNA has been shown to reduce pathology and improve cognition in animal models and is now being studied in ALS patients with the C9orf72 repeat expansion. Reference Jiang, Zhu and Gendron112
Conclusion
The FTD field is currently undergoing a paradigm shift, with recent advances in our understanding of the molecular pathogenesis finally making disease-modifying treatments a realistic possibility. However, the testing and implementation of these targeted therapies will require further improvements in early, accurate clinical diagnosis and the development of biomarkers that allow for better classification of patients so they can be directed towards the most appropriate therapy.
Funding
The authors have not received any funding to support the present manuscript. During the past 36 months, the authors have received funding from CIHR (IM, CM), NIH (IM, CM), Weston Brain Institute (IM), ALS Canada (IM), Alzheimer’s Association US (IM), Biogen (CM), Avanex (CM), Janssen (CM) and Green Valley (CM).
Conflicts of Interest
IM has received licensing fees from Prevail Therapeutics related to US patent 12/302691 and support to attend the PLS Conference and annual meeting of the CANP. CT Has a leadership role on boards of the PSP Society of Canada, Alzheimer’s Society of Toronto, Women’s Brain Project and Brain Injury Canada.
Statement of Authorship
MT and IM wrote and edited the manuscript.







 Open access
Open access