



N-methyl-D-aspartate receptor (NMDA) encephalitis is a recently described autoimmune disease that typically presents with prodromal symptoms including upper respiratory tract infection, headache, fever, nausea, vomiting and diarrhea. Psychiatric symptoms follow within weeks, including anxiety, insomnia, mania, paranoia and grandiose delusions. The diagnosis is confirmed by the detection of NMDA antibodies in the serum or cerebrospinal fluid (CSF). Reference Dalmau, Lancaster, Martinez-Hernandez, Rosenfeld and Balice-Gordon1 Tumours, especially teratomas, are frequently associated with NMDA encephalitis; however, only 5% of male patients older than 18 years have been found to have an underlying tumour. Optic neuropathy associated with NMDA encephalitis is being increasingly recognised in the literature Reference Mugavin, Mueller, Desai and Golnik2 – Reference Motoyama, Shiraishi, Tanaka, Kinoshita and Tanaka6 and was reviewed most recently by Mugavin et al. Reference Mugavin, Mueller, Desai and Golnik2 in 2017. In this report, we present a case of bilateral optic neuropathy in a young man diagnosed with NMDA receptor encephalitis.
A 25-year-old male of South Asian descent presented with acute confusion, ataxia and hallucinations as well as fever, nausea and vomiting. On examination, pupils were equal and reactive to light with full extraocular movements, but further ophthalmic examination was not completed due to lack of cooperation. He declined to read the eye chart, but did exhibit normal visual behaviour. As per his family, he had not described vision loss, had no past ocular or medical history and was on no medications.
He was admitted for presumed viral encephalitis but quickly deteriorated and required transfer to the intensive care unit. He developed seizures which progressed to refractory status epilepticus with bilateral anterior-temporal foci.
Infectious workup of blood and CSF was positive for respiratory syncytial virus (RSV) subgroup A but negative for herpes simplex, varicella zoster, tuberculosis, HIV, arbovirus, legionella, lyme, syphilis, hepatitis A, B and C, toxoplasmosis, malaria and West Nile virus. Paraneoplastic antibodies (anti-Hu, anti-Yo, anti-Ri, anti-Ma:Ta, anti-CV2, anti-Amphiphysin and anti-GAD5) were negative. Opening pressure was not measured. CT head, chest, abdomen and pelvis were all normal as was a scrotal ultrasound. Baseline magnetic resonance imaging (MRI) demonstrated nonspecific punctate foci of T2-weighted (T2W) Fluid-Attenuated Inversion Recovery signal hyperintensity, in the cerebral white matter with normal cerebral morphology and normal appearance of the optic nerves (Figure 1a).
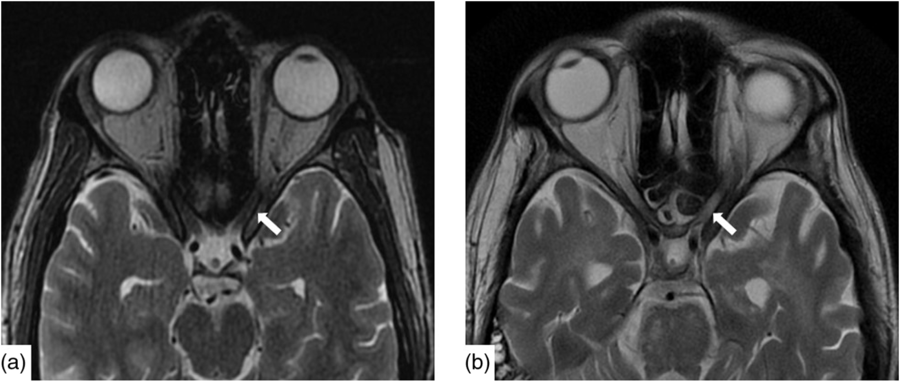
Figure 1: Axial T2W imaging demonstrated baseline (a) normal signal intensity of the canalicular segment of the optic nerves. Within 2 months, (b) there is new, notable signal hyperintensity in the optic nerves bilaterally.
Due to lack of improvement, he was empirically treated with high-dose steroids and intravenous immunoglobulin for possible autoimmune disease, at which time CSF testing for anti-NMDA returned positive. He was treated with two doses of cyclophosphamide 1290 mg and four doses of rituximab 645 mg.
Nineteen days after admission pupils became sluggish, then 2 weeks later, became non-reactive to light with absent corneal and gag reflexes. Two months after presentation, he remained intubated and no objective determination of visual function could be obtained. Both pupils were fixed and unreactive to light. He also had bilateral lagophthalmos, severe dry eye and filamentary keratitis. Both optic discs were pale with circumpapillary high water marks suggestive of resolving optic disc oedema. In the left eye, there were flat white peripapillary retinal lesions extending into the macula, consistent with retinal nerve fibre layer infarcts. Repeat MRI demonstrated striking new T2W hyperintensity in the optic nerves (Figure 1b). Over 4 months, the retinal lesions gradually regressed followed by the high water marks, but the discs remained pale. The filamentary keratitis improved with aggressive lubrication and as he regained consciousness and the ability to blink. MRI 6 months after presentation revealed severe, global volume loss, including striking atrophy of the optic chiasm. There was also confluent, extensive signal abnormality throughout much of the brain (Figure 2). The patient was improving clinically but unexpectedly died 10 months after admission to hospital from respiratory complications.
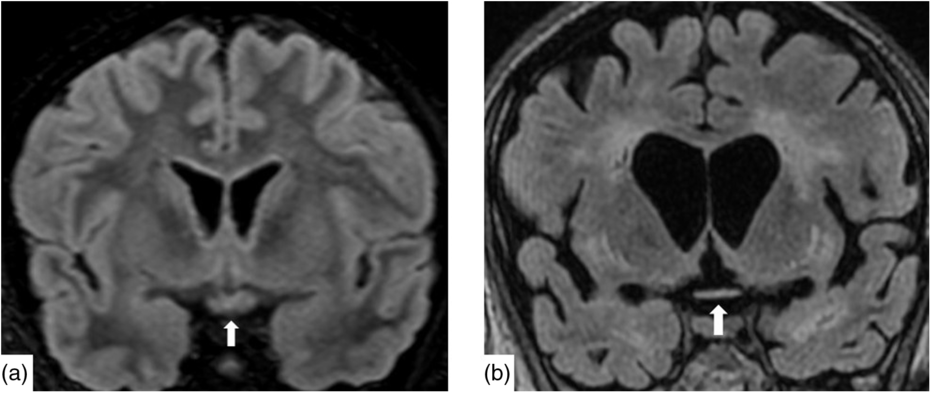
Figure 2: Coronal reconstruction of a volumetric Fluid-Attenuated Inversion Recovery (a) demonstrating normal signal and morphology of the optic chiasm. Contrast with 6 months after presentation (b) demonstrating severe atrophy of the chiasm and global volume loss. There is new severe white matter signal change in much of the imaged brain.
Of the seven reported cases of optic neuropathy associated with NMDA receptor encephalitis, most initially presented as optic neuritis Reference Mugavin, Mueller, Desai and Golnik2 , Reference Zoccarto, Saddi and Serra4 – Reference Motoyama, Shiraishi, Tanaka, Kinoshita and Tanaka6 and one developed aquaporin-4-antibody positive optic neuritis 9 months after the encephalitis. Reference Yang, Kim, Park, Park, Kim and Hwang3 In our case, there were no known visual complaints upon presentation, after which the patient had a rapid decrease in his level of consciousness. The gradual loss of pupillary response prompted an ophthalmology consultation only 2 months after presentation, at which time optic atrophy had already occurred. The presence of retinal nerve fibre layer infarcts and high water marks were suggestive of resolved optic disc oedema. Although opening pressure was not measured, there were no radiographic features suggestive of elevated intracranial pressure, making papilledema an unlikely cause of optic disc swelling. Bilateral optic nerve T2 hyperintensity on MRI was consistent with the diagnosis of optic neuritis.
The differential diagnosis of optic disc pallor is broad, and NMDA receptor encephalitis is an uncommon cause of optic neuropathy. Our patient demonstrated marked atrophy of the anterior visual pathway over the course of his illness, confirming that optic atrophy occurred simultaneously with the encephalitis. He underwent extensive testing as part of the initial workup, and no other known cause of optic atrophy was found. The first case of a patient developing neuromyelitis optica following NMDA receptor encephalitis was reported in 2013, suggesting a common inflammatory pathway. Reference Zoccarto, Saddi and Serra4 There now appears to be a link between NMDA receptor encephalitis and demyelinating disease in 5% of patients. Reference Dalmau, Armangué and Planagumà7 Our patient represents an additional case of inflammatory optic neuropathy associated with NMDA receptor encephalitis.
In our case, profound encephalopathy and resultant decreased level of consciousness prevented any formal assessment of vision, or even subjective complaints of vision loss. We postulate that it is possible that optic neuropathy is more common in this disease than that has been reported, since acutely psychotic patients may not complain of blurred vision. Furthermore, the high mortality rate makes follow-up assessment challenging. Reference Dalmau, Lancaster, Martinez-Hernandez, Rosenfeld and Balice-Gordon1 , Reference Mugavin, Mueller, Desai and Golnik2
The link between NMDA receptor encephalitis and optic neuropathy has not been widely reported and remains preliminary, but it is possible these two are part of the same autoimmune syndrome. Reference Colley and Smith5 The goal of this case report is to provide additional evidence for the possible link between these two entities and to demonstrate changes seen on neuroimaging. The progressive atrophy on MRI of the anterior visual pathway has not previously been reported. Given its increasing frequency in the literature, reviewing imaging changes in the optic pathway in future patients could provide additional evidence linking optic neuropathy and NMDA receptor encephalitis, especially in patients who are unable to articulate changes in their vision.
Disclosures
The authors have nothing to disclose.
Statement of Authorship
DCS: Manuscript writing and editing. SKP: Manuscript writing and figure preparation. LLCDB: Manuscript writing and editing.


