Limb-girdle muscular dystrophies (LGMD) are a heterogeneous group of inherited myopathies with a prevalence of approximately 2 per 100,000. Reference Norwood, Harling, Chinnery, Eagle, Bushby and Straub1 LGMD recessive 1 (LGMDR1) is the most common subtype of LGMD. It is a recessive disorder caused by mutations in calpain 3 (CAPN3). Intronic mutations that affect splicing and other non-coding variants comprise at least 20% of the >490 mutations reported to date. Reference Fanin and Angelini2 This figure likely represents an underestimate, as mutations in non-coding sequences may be missed by exon-based sequencing.
The use of next-generation sequencing in cohorts of myopathy patients has led to the identification of single heterozygous variants in many genes known to cause recessive LGMDs. Reference Savarese, Di Fruscio and Torella3 This finding highlights the need for an additional technique to identify coexisting non-coding mutations that may be causative of these patients’ myopathies. Sanger sequencing of cDNA has previously been used to identify mutations affecting CAPN3 splicing in some such patients. Reference Duno, Sveen, Schwartz and Vissing4 Other approaches exist but carry limitations. Western blot has limited sensitivity and specificity. Reference Fanin and Angelini2 Assays of calpain 3 proteolytic function are not widely available, Reference Fanin and Angelini2 nor are functional assays for most other proteins involved in inherited myopathies. RNA-sequencing has the potential to overcome these limitations by allowing the simultaneous assessment of variants, aberrant splicing and expression of many genes. Recently, this technique was used to identify new pathogenic variants in patients with various inherited myopathies, muscular dystrophies and spinal muscular atrophy. Reference Cummings, Marshall and Tukiainen5–Reference Hamanaka, Miyatake and Koshimizu7 Variants identified by RNA-sequencing in NEB and COL6A1 proved to be common but previously unrecognised causes of nemaline and collagen VI-related myopathies. We report here a case of LGMDR1 carrying a novel splice site variant in CAPN3 identified by RNA-sequencing.
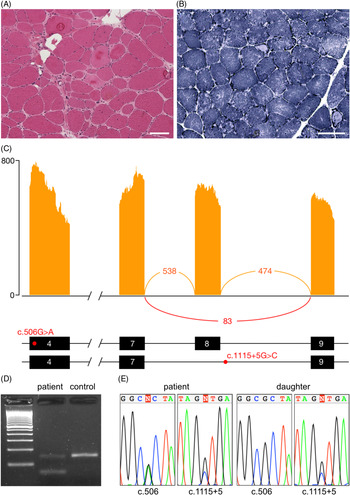
The proband is a 68-year-old woman of Colombian descent with no family history of neuromuscular disease. She first presented to medical attention at age 39 with shoulder pain and exercise intolerance. Over the course of the following 5 years, she developed weakness affecting the proximal upper and lower limbs, as well as scapular winging. The weakness continued to progress over the subsequent years, with the patient ultimately requiring a wheelchair by age 56. Pulmonary function tests, an electrocardiogram and an echocardiogram were all within normal limits. Creatine kinase levels were elevated, up to 4059 IU/L (normal 15–200 IU/L). An electromyogram showed myopathic findings with fibrillation potentials. A muscle biopsy showed dystrophic findings and a large number of lobulated fibres (Figure 1(A) and (B)).

Figure 1: Muscle histology and aberrant splicing of CAPN3 in an LGMDR1 patient. (A) Haematoxylin and eosin stained muscle tissue showing dystrophic changes, including variation in fibre size, hypertrophic fibres, fibre splitting, necrotic and regenerating fibres, internalised nuclei and endomysial fibrosis. (B) Nicotinamide adenine dinucleotide (NADH) staining showing moth-eaten and lobulated fibres. Scale bar 100 μm in all panels. (C) Sashimi plot from RNA-seq illustrating the skipping of exon 8, as well as the position of the c.506A>G and c.1115+5G>C variants within the transcript. (D) Validation of the alternative splicing by RT-PCR. The lower band in the patient corresponds to the transcript lacking exon 8. (E) Chromatograms demonstrating presence of the c.506A>G and c.1115+5G>C variants in the patient and c.1115+5G>C in her unaffected daughter.
DNA-based panel sequencing of 176 myopathy genes (MNG Laboratories, Atlanta, GA, USA) identified heterozygous variants of unknown significance in five genes, two of which had previously been associated with an LGMD phenotype. The first of these was a missense variant (c.506G>A; p. Arg169His) in exon 4 of CAPN3 (NM_000070). The second was a missense variant (c.689G>T; p. Gly230Val) in ANO5 (NM_00114264). The latter was previously reported to cause LGMDR12 but is also found in the homozygous state in one individual in the gnomAD database (gnomad.broadinstitute.org). The c.506G>A variant is located in the proteolytic domain of calpain 3. It is present at a low frequency (0.001%) in the gnomAD database but had not previously been reported as causative of LGMDR1. It was predicted to be deleterious by the SIFT, PolyPhen2, MutationTaster and combined annotation dependent depletion algorithms (CADD). Variants affecting nearby residues (p.167, p.168 and p.170) have been reported as pathogenic or likely pathogenic. Based on these parameters, this variant was classified as likely pathogenic according to American College of Medical Genetics guidelines.
This study was approved by the McGill University Health Centre Research Ethics Board. All participants provided informed consent. RNA was extracted from snap-frozen muscle tissue. Library generation was performed using the TruSeq stranded mRNA library preparation kit (Illumina, San Diego, CA, USA), and sequencing was performed on an Illumina HiSeq 2500 using 125 bp paired-end reads and 0.25 lanes of data per sample. Alignment was performed using STAR against a reference genome (Hg19). Variant calling was performed using GATk. Splicing variants were identified using the JSplice and cufflinks software packages, as well as manual review of selected genes. Read counts were obtained using featureCounts, and gene expression was quantified using DESeq2.
On RNA-sequencing, an allelic imbalance was observed for the c.506G>A variant, with the alternative A allele representing 87% of reads, suggesting degradation and nonsense-mediated decay of the second allele. This hypothesis was supported by decreased expression of CAPN3 transcripts compared to two normal controls (log2 fold change: −1.58, p-value: 0.0003). Western blot was performed in a clinical laboratory and showed a protein of normal size; protein expression was not quantified. Aberrant splicing analysis identified CAPN3 transcripts lacking exon 8, leading to a frameshift and a premature stop codon (Figure 1(C)). This splicing variant was confirmed by RT-PCR (Figure 1(D)). Sanger sequencing of the boundaries of exon 8 identified a novel c.1115+5G>C variant (Figure 1(E)). The c.1115+5G>T variant at the same position has previously been reported to be pathogenic. Reference Chae, Minami and Jin8 Testing in the patient’s unaffected daughter confirmed that the missense and splice variants are in trans. No aberrant splicing was identified in any other myopathy genes.
In this report, we describe the combination of gene panel and RNA-sequencing to establish a molecular diagnosis in an LGMDR1 patient, by identifying two variants in CAPN3. One of these variants is an intronic variant leading to skipping of exon 8 and was only identified after RNA-sequencing demonstrated transcripts lacking this exon. While such variants are sometimes identified by DNA sequencing, their functional effects are often unknown, and many commercial laboratories thus only report variants within the canonical splice site itself, as occurred in our patient. This case illustrates the importance of mutations in non-coding sequences as a likely under-diagnosed cause of LGMDR1 and other inherited myopathies. It also illustrates the utility of RNA-sequencing as a second-line diagnostic modality when panel or exome sequencing is negative, or when it reveals only a single heterozygous variant in a gene known to cause a recessive disorder. This technique is of particular relevance in myopathies in which non-coding mutations are estimated to account for a significant proportion of cases, such as calpainopathies and dysferlinopathies. A proposed diagnostic algorithm that incorporates RNA-sequencing is outlined in Figure 2.

Figure 2: Proposed algorithm for the molecular diagnosis of LGMDs. * Targeted testing for disorders not amenable to diagnosis by next-generation sequencing (e.g. facioscapulohumeral muscular dystrophy, myotonic dystrophy type 2). LGMD = limb girdle muscular dystrophy; RNA-seq = RNA sequencing; VUS = variant of unknown significance; WES = whole exome sequencing; WGS = whole genome sequencing.
In the investigation of undiagnosed Mendelian disorders, RNA-sequencing provides information that is complementary to DNA sequencing, chiefly by evaluating the impact of variants on splicing. RNA-sequencing can also identify certain rearrangements such as inversions, Reference Cummings, Marshall and Tukiainen5 which are not amenable to detection by exome sequencing. Finally, while some of this information can be gathered through targeted sequencing of the transcripts of a single gene, this technique is impractical when there are multiple candidate genes, as was the case in our patient. While RNA-sequencing can be performed from blood, Reference Kernohan, Fresard and Zappala6 many of the genes and isoforms involved in muscle disease are poorly expressed in leukocytes. Reference Cummings, Marshall and Tukiainen5 Sequencing from muscle tissue is therefore preferable, potentially posing an obstacle to use of RNA-sequencing in some cases. In summary, this report highlights the value of RNA-sequencing as a diagnostic tool in inherited muscle disease, as it can aid in the identification of new pathogenic variants in non-coding sequences of known disease genes.
Acknowledgements
We would like to thank the patient who participated in this study. This work was made possible by funding from the Fondation du Grand Défi Pierre Lavoie, the McGill University department of Pathology, the McGill University Health Centre Research Institute, the Montreal Neurological Institute, and the Fonds de recherche du Québec – Santé (FRQS). MT received a Junior 1 salary award from the FRQS. KC received a doctoral award from the FRQS.
Disclosures
Dr. O’Ferrall reports personal fees from PTC Therapeutics nmDMD Advisory board, grants from Sanofi Genzyme, grants from Acceleron, grants from Grifols, personal fees from Biogen, outside the submitted work. The other authors have no conflicts of interest to declare.
Statement of Authorship
SN reviewed clinical data, participated in the RNA-sequencing data analysis, conducted the validation experiments and analysis, and wrote the manuscript. KC prepared the samples and revised the manuscript. EB developed the bioinformatics pipeline, produced the RNA-sequencing data and revised the manuscript. YHS performed the RNA extraction. BB reviewed the clinical data and revised the manuscript. EO performed the clinical evaluation, performed the biopsy and revised the manuscript. MT planned the study, analysed the RNAseq data, analysed the validation data and wrote the manuscript. JK planned the study, performed the pathological evaluation and revised the manuscript.