


A 48-year-old Polish man was evaluated for difficulties in climbing stairs and holding his upper limbs above the head level, which started at the age of 30 and progressively worsened. He also complained of breathing difficulties while performing light physical activities. His medical history was otherwise unremarkable, and no other member of his family suffered from neurological diseases. Physical examination showed upper and lower limb proximal weakness, normal muscle tone, deep tendon reflexes, and plantar responses; sensory examination was normal. Creatine kinase level was three times above the normal value (596 UI/l; range, 30-170). Needle electromyography in the deltoid, biceps brachii, and quadriceps showed a mixed pattern with early, myopathic recruitment (small-amplitude polyphasic motor unit potentials with interference reached for low effort), associated with rare high-amplitude motor unit potentials superimposed at maximal effort, fasciculations (high-amplitude triphasic spikes, firing inconstantly at approximately 1/second) and sporadic fibrillation potentials at rest. Motor and sensory nerve conduction studies in the upper and lower limbs were normal except for a slightly decreased amplitude of the sural nerve sensory action potential, which had a normal velocity. Mild respiratory failure with reduction of forced vital capacity at 50% of predicted value was documented by pulmonary function tests.
To further address the differential diagnosis between a primary muscle disease, supported by the early full recruitment, and a lower motor neuron disorder, supported by the presence of signs compatible with denervation such as fibrillation and fasciculation potentials and high-amplitude motor unit potentials, additional diagnostic tests were performed. These included magnetic resonance imaging (MRI) of the lumbosacral spine that excluded radiculopathies and lower limb muscle MRI that showed severe fatty replacement of gluteus medius, adductor longus, and to a lesser extent semimembranosus, together with increased signal intensity on short-tau-inversion-recovery sequences in several thigh muscles (Figure 1A, B). A muscle biopsy of the rectus femoris under local anesthesia with lidocaine was requested; just before the muscle specimen withdrawal, a remarkable and continuous fasciculation activity was noticed through the surgical incision (Online Video 1). Muscle pathology was suggestive of glycogen storage disease associated with scattered or grouped angulated fibers in all fascicles (Figure 1C-E) without type grouping. Enzymatic acid alpha-glucosidase activity on muscle sample was markedly reduced (4% of normal value) and a diagnosis of glycogenosis type II (GSD II) was made. Enzyme replacement therapy was not started because the patient went back to his country of origin and is not currently followed at our Institution.
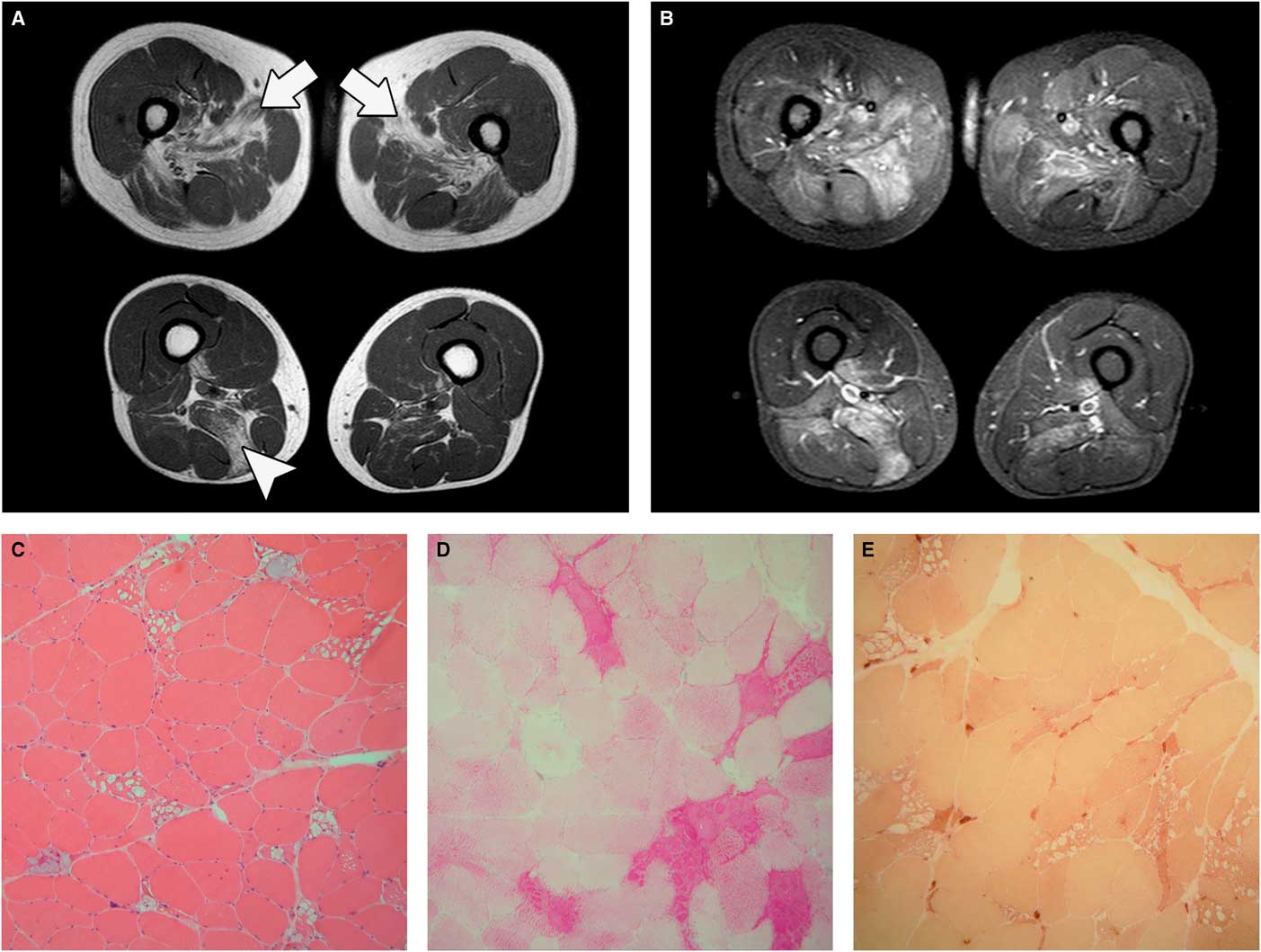
Figure 1 Muscle MRI. (A) Axial T1-weighted sequences showing adductor longus (arrows) and semimembranosus (arrowhead) fatty replacement. (B) Axial short-tau-inversion-recovery (STIR) sequences showing muscle hyperintensity. Muscle pathology, 20× magnification: (C) hematoxylin-eosin; (D) periodic acid–Schiff (PAS) staining; and (E) nonspecific esterase. Sample taken from fasciculating muscle shows the presence of many angulated fibers next to fibers bearing large cytoplasmic vacuoles full of PAS-positive material.
GSD II, also known as Pompe disease or acid maltase deficiency, is a lysosomal glycogen storage disease caused by acid alpha-glucosidase deficiency.Reference Van der Ploeg and Reuser 1 Two main clinical subtypes are recognized: (1) the infantile-onset form, characterized by a severe cardiac, motor, and respiratory impairment that usually appears during the first months of life; and (2) the adult- or late-onset form, frequently resembling a limb-girdle muscular dystrophy in the pattern of muscle weakness, with involvement of the diaphragm, intercostal and accessory muscles leading to respiratory failure.Reference Lamperti, Crugnola, Comi and Moggio 2 In our case, muscle imaging showing a predominant involvement of adductor longus and semimembranosus muscles together with short-tau-inversion-recovery hyperintensities was compatible with late-onset Pompe disease,Reference Díaz-Manera, Llauger, Gallardo and Illa 3 although other differential diagnoses such as limb-girdle muscular dystrophies or even facioscapulohumeral muscular dystrophyReference Tasca, Monforte and Ottaviani 4 could not be completely ruled out just based on thigh MRI findings.
Recent evidence suggest that glycogen accumulation may occur in several other tissues besides the heart and skeletal muscle, such as the liver, kidney, spleen, salivary glands, glial cells, brainstem nuclei, anterior horn cells of spinal cord, and smooth muscle in GSD II, supporting that it should be considered as a multisystemic disorder.Reference Filosto, Todeschini and Cotelli 5 The involvement of both central and peripheral neurons is well-documented in animal models and in the infantile forms of the disease,Reference DeRuisseau, Fuller and Qiu 6 whereas data available for the adult-onset form are scarce.
Fasciculations arise as a result of spontaneous depolarization of lower motor neurons leading to the synchronous contraction of all the muscle fibers within a single motor unit. They can be found in different disorders affecting motor neurons or peripheral nerves or may not even be associated with disease in the so-called benign fasciculation syndrome. The coexistence of a motor neuropathy/radiculopathy might have been a possible explanation for their presence in our patient, but the investigations performed did not support this hypothesis. Anterior horn cell damage resulting from glycogen accumulation in GSD II, indirectly supported by the presence of denervated fibers on muscle biopsy (Figure 1E), may be the cause of the significant fasciculations in our patient. Indeed, esterase-positive angulated atrophic fibers have already been reported in juvenile/adult GSD II biopsiesReference Werneck, Lorenzoni, Kay and Scola 7 as well as electromyography findings of spontaneous activity and prolonged high-amplitude motor unit potentials.Reference Hobson-Webb, Dearmey and Kishnani 8 , Reference Müller-Felber, Horvath and Gempel 9 Clinical, electrophysiological, and pathological findings of motor neuron involvement in adult GSD II patients could be, in the absence of other evident causes, related to glycogen storage in motor neurons.
Disclosures
MM, SS, ER, and GT do not have anything to disclose.
Statement of Authorship
MM and GT collected and analyzed data and drafted the manuscript; SS and ER collected data and reviewed the manuscript for intellectual content.
SUPPLEMENTARY MATERIAL
To view supplementary material for this article, please visit https://doi.org/cjn.2016.447


