Introduction
Mitochondrial disorders (MIDs) are usually multisystem diseases (mitochondrial multiorgan disorder syndrome [MIMODS]), either already at onset or with progression of the disease.Reference Finsterer and Zarrouk-Mahjoub 1 One of the organs most frequently involved in MIDs is the brain.Reference Finsterer 2 Cerebral manifestations in MIDs are variable and may be classified as pure clinical without abnormalities on imaging or functional studies, as clinical with functional or imaging abnormalities, or as functional or imaging abnormalities without appropriate clinical manifestations (Table 1).Reference Finsterer 2 This review aims at summarizing and discussing recent findings and future perspectives concerning the clinical presentation, pathophysiology, diagnosis, treatment, and outcome of cerebral disease in MIDs.
Table 1 Classification of CNS abnormalities in MIDs according to the predominant presentation either on clinical examination or on imaging/functional studies
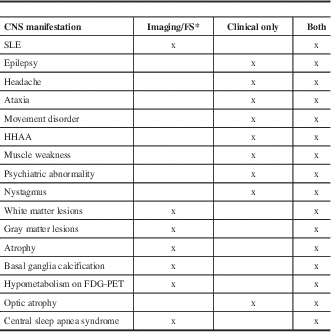
*Instrumental investigations are inevitable for diagnosing stroke-like episodes (SLEs), gray matter lesions, white matter lesions (WMLs), cerebral atrophy, basal ganglia calcification, hypometabolism, and sleep apnea syndrome.
FS=functional studies, HHAA = hypothalamic-hypophysial-adrenal axis.
Classification
Cerebral abnormalities in MIDs may not only be classified as pure clinical (e.g. headache) or as clinical with abnormalities on functional or imaging studies (e.g. stroke-like episode [SLE]) but, depending on the affected tissue, also as vascular, astrocytic, or neuronal. Cerebral manifestations of MIDs may be permanent (e.g. dementia) or transient (e.g. seizure, SLE, headache) and may be a direct consequence of the metabolic defect (e.g. SLE) or secondary resulting from involvement of other organs (e.g. stroke from atrial fibrillation, bleeding from hypertension). Central nervous system (CNS) abnormalities of MIDs may be also categorized as treatable (e.g. epilepsy) or inaccessible to treatment (e.g. basal ganglia calcification, atrophy). Additionally, a CNS abnormality may go along with or without other CNS abnormalities attributable to the MID. Furthermore, cerebral abnormalities in MIDs may or may not be accompanied by manifestations in other organs (MIMODS). CNS involvement in MIDs may be also categorized according to the affected anatomical structure (cortex, subcortical, white matter, basal ganglia, thalamus, midbrain, pons, cerebellum, medulla, or spinal cord) or according to the onset of the clinical manifestations as early or late onset. Finally, cerebral lesions can be delineated from spinal cord lesions and CNS lesions resulting from respiratory-chain defects can be delineated from CNS lesions from nonrespiratory chain mitochondrial defects.
CNS Manifestations of MIDs
There are several clinical CNS abnormalities with or without concomitant morphological/functional abnormalities and several morphological and functional abnormalities with or without clinical manifestations, which have been identified as manifestations of specific and nonspecific MIDs (nsMIDs) (Table 1). These include SLEs, epilepsy, headache, ataxia, movement disorders, nystagmus, muscle weakness, insufficiency of the hypothalamic-hypopituitary-adrenal axis, muscle weakness, psychiatric abnormalities, nystagmus, white matter lesions (WMLs), gray matter lesions, atrophy, basal ganglia calcification, and hypometabolism on 2-deoxy-2-[fluorine-18]fluoro-D-glucose positron-emission tomography (FDG-PET) (Table 1).
SLEs
SLEs are a typical phenotypic feature of mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS) syndrome, with which they occur in the majority of the patients. However, SLEs have been also reported in patients with myoclonus epilepsy with ragged-red fibers (MERRF) syndrome,Reference Vastagh, Gál and Reményi 4 Kearns-Sayre syndrome (KSS),Reference Furuya, Sugimura, Yamada, Hayashi and Kobayashi 5 Saguenay-Lac-St. Jean cytochrome oxidase deficiency,Reference Morin, Dubé and Robinson 6 Leigh syndrome,Reference Matsui, Takano and Ryujin 7 and coenzyme-Q deficiency resulting from ADCK3 mutations.Reference Hikmat, Tzoulis and Knappskog 8 Additionally, SLEs have been reported in nonmitochondrial conditions, such as X-linked hereditary motor and sensory neuropathy (HMSN1),Reference Anand, Maheshwari and Roberts 9 neurobrucellosis,Reference Reggio, Vinciguerra, Sciacca, Fiumanò, Iacobello and Zappia 10 , cerebral amyloid angiopathy,Reference Mendonça, Caetano, Pinto and Cruz e Silva 11 or sarcoidosis.Reference Campbell, Kee, Bhattacharya, Flynn, McCarron and Fulton 12 Clinical presentation of SLEs can be heterogeneous. The most frequent symptom of an SLE is cortical blindness.Reference Ichikawa 3 Other clinical manifestations include psychiatric disorders,Reference Wang and Le 13 epilepsy,Reference Fryer, Bain and De Vivo 14 headache,Reference Tsujikawa, Yokoi, Yasui, Hasegawa, Hoshiyama and Yanagi 15 hemiparesis,Reference Mordaunt, McIntyre and Salvemini 16 and various types of aphasia.Reference Namer, Wolff, Dietemann and Marescaux 17 More rarely, visual agnosia, prosopagnosia, cortical deafness, auditory agnosia (from the mutation m.10197G>A), topographical disorientation, disinhibition, agitation, euphoria, anxiety, impaired face recognition, prolonged visual aura, hemianopia or quadrantanopia, or hemispatial neglect have been reported.Reference Ichikawa 3 , Reference Namer, Wolff, Dietemann and Marescaux 17 , Reference Jung, Park and Kim 18
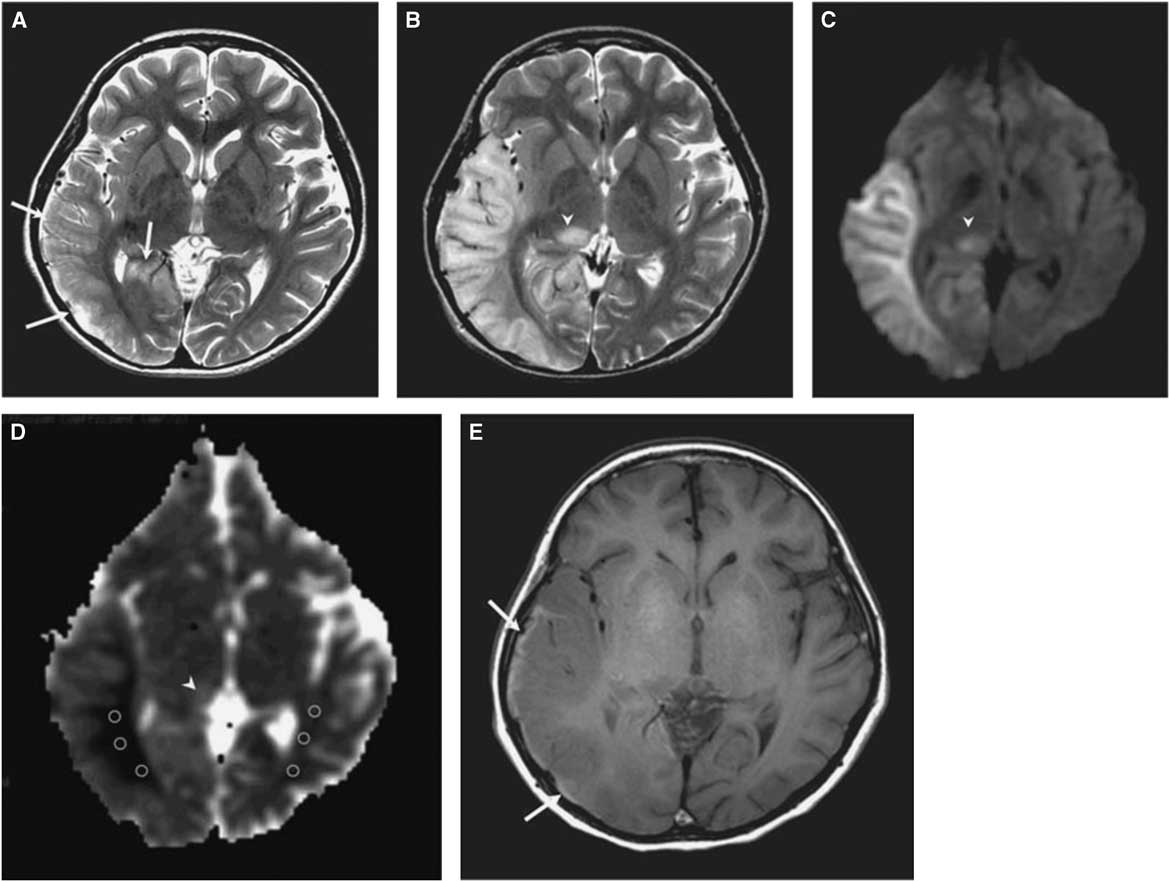
The morphological correlate of an SLE on cerebral imaging is the stroke-like lesion (SLL). Depending on the interval after onset, an acute or chronic stage of an SLL can be delineated. The acute stage of an SLL on magnetic resonance imaging (MRI) is characterized by hyperintensity on T2-w/fluid-attenuated inversion recovery images, hyperintensity on diffusion weighted imaging (DWIs), and hyperintensity on apparent diffusion coefficient (ADC) maps (Figure 1). Occasionally, areas with cytotoxic edema within the SLL may be found. Blood flow is increased on perfusion weighted imaging in the acute stage. Magnetic resonance spectroscopy may show a lactate peak and a reduced N-acetyl-aspartate/creatine ratio indicating neuronal death (Table 2).Reference Wang, Xiao, Xie, Zhao, Liu, Zhang, Yuan and Huang 19 , Reference Finsterer 20 A lactate peak is regarded as abnormal only if the N-acetyl-aspartate/choline ratio is normal. In a study of 13 patients with, altogether, 44 SLLs, DWI showed hyperintensity in 37 and isointensity in seven cases.Reference Kim, Lim and Jeon 21 On ADC, 16 were hyperintense, 16 hypointense, and 15 isointense.Reference Kim, Lim and Jeon 21 The chronic stage of SLLs is characterized by spreading and later regression of the lesion, hyperintensity, hypointensity, or isointensity on T2,Reference Kim, Lim and Jeon 21 hyperintensity, fainting or disappearance on DWI, hypointensity or isointensity on ADC, and hypoperfusion.Reference Wang, Xiao, Xie, Zhao, Liu, Zhang, Yuan and Huang 19 Outcomes from SLLs include complete recovery, focal atrophy, laminar cortical necrosis, or a WML.Reference Kim, Lim and Jeon 21 , Reference Renard and Taieb 22 Besides SLEs, patients with MIDs may experience ordinary ischemic strokes or transitory ischemic attacks secondary to cardiac involvement in the MID.Reference Mitani, Aida, Tomiyasu, Wada and Osaka 23 SLEs are frequently accessible to the nitric oxide precursors L-arginine (500 mg/kg/d), citrulline, or succinate. Supportive measures include a ketogenic dietReference Steriade, Andrade, Faghfoury, Tarnopolsky and Tai 24 and symptomatic treatment of the various clinical manifestations of an SLE.Reference Finsterer and Bindu 25
Table 2 Specific and nonspecific MIDs with CNS involvement and location of the predominant genetic defect
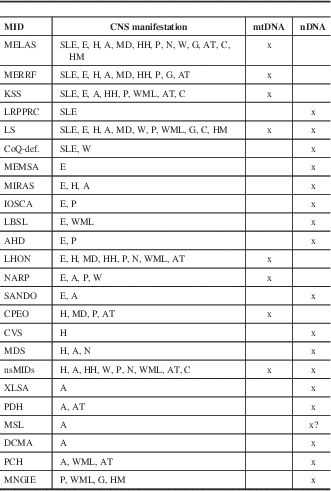
?=uncertain, A=ataxia, AT=cerebral atrophy, C=basal ganglia calcification, CoQ-def = coenzyme Q deficiency, DCMA = dilated cardiomyopathy with ataxia, E=epilepsy, G=gray matter lesions, H=headache, HH=hypothalamic-hypophysial axis, HM=hypometabolism, IOSCA=infantile onset spinocerebellar ataxia, LBSL=leukoencephalopathy, brainstem and spinal cord lesions, and lactic acidosis, MD=movement disorder, MEMSA=myoclonic epilepsy myopathy sensory ataxia, MSL=multiple systemic lipomatosis, N=nystagmus, P=psychiatric abnormalities, SANDO=sensory ataxic neuropathy, dysarthria, and ophthalmoparesis, W=muscle weakness or hypotonia, WML=white matter lesions
Epilepsy
Mitochondrial epilepsy is a common feature of specific and nsMIDs. Epilepsy may be the dominant feature (e.g. MERRF) or nondominant feature (e.g. Leber hereditary optic neuropathy (LHON)) of the phenotype. All types of seizures may occur with mitochondrial epilepsy, but focal seizures appear more frequent than generalized seizures. However, no systematic studies on this matter have been carried out. According to a literature review, focal seizures with secondary generalization were more prevalent than primary generalized seizures in pediatric MIDs, which are more frequently the result of nuclear DNA than mitochondrial DNA (mtDNA) mutations.Reference Finsterer and Zarrouk Mahjoub 26 In adult MIDs, which are more frequently from mtDNA than nuclear DNA mutations, generalized seizures are more prevalent than focal seizures.Reference Finsterer and Zarrouk Mahjoub 26 A common type of epilepsy in MIDs is myoclonic epilepsy. Among the specific MIDs, mitochondrial epilepsy with early onset occurs in MELAS, MERRF, KSS, Leigh syndrome, myoclonic epilepsy myopathy sensory ataxia, mitochondrial recessive ataxia syndrome (MIRAS), infantile onset spinocerebellar ataxia (IOSCA), leukoencephalopathy, brainstem and spinal cord lesions, and lactic acidosis, and Alpers-Huttenlocher syndrome.Reference Finsterer and Zarrouk Mahjoub 26 Mitochondrial epilepsy with adult onset has been reported in MELAS, LHON, neuropathy ataxia and retinitis pigmentosa (NARP), and sensory ataxic neuropathy, dysarthria, and ophthalmoparesis.Reference Finsterer and Zarrouk Mahjoub 26 In a study of seven MELAS patients, seizures usually occurred during the acute phase of an SLE and included epilepsia partialis continua, hemiclonic status epilepticus, nonconvulsive status, and occipital status epilepticus.Reference Demarest, Whitehead, Turnacioglu, Pearl and Gropman 27 Among pediatric patients, infantile spasms, refractory or recurrent status epilepticus, epilepsia partialis continua, and myoclonic epilepsy were the most prevalent seizure types.Reference Desguerre, Hully, Rio and Nabbout 28 In a retrospective study of 109 pediatric and adult MID patients undergoing electroencephalography, 85% had epileptiform discharges, including multifocal discharges (41%), focal discharges (39%), and generalized discharges (39%).Reference Chevallier, Von Allmen and Koenig 29 The most common types of seizures were complex partial (37%) and generalized tonic-clonic (39%).Reference Chevallier, Von Allmen and Koenig 29 Among those with seizures (55%), 28% were intractable to treatment.Reference Chevallier, Von Allmen and Koenig 29 Patients with Leigh syndrome most commonly had focal or generalized seizures (11% in both) and patients with MELAS most commonly had generalized seizures (33%).Reference Chevallier, Von Allmen and Koenig 29 NARP may be associated with catastrophic epilepsy.Reference Keränen and Kuusisto 30 Intractable seizures with epileptic encephalopathy have been also reported in patients carrying CARS2 mutations associated with combined respiratory chain deficiency of complexes I, III, and IV (Table 3).Reference Coughlin, Scharer and Friederich 31
Table 3 Respiratory chain defects in MIDs with CNS involvement
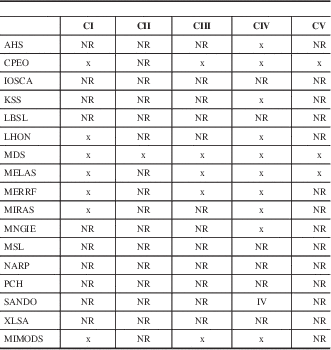
AHS=Alpers-Huttenlocher syndrome, NR=not reported.
Treatment of mitochondrial epilepsy mainly relies on antiepileptic drugs (AEDs). Additional measures include epilepsy surgery, diets, vagal nerve stimulation, and supportive agents.Reference Finsterer and Zarrouk-Mahjoub 32 Treatment should start with AEDs with a low mitochondrion-toxic potential, such as levetiracetam, lamotrigine, gabapentin, or zonisamide. Only when these agents are ineffective or accompanied by severe side effects should AEDs with high mitochondrion-toxic potential, such as valproic acid, carbamazepine, phenytoin, or phenobarbital, be tried.Reference Finsterer and Zarrouk-Mahjoub 32 Valproic acid seems to have one of the highest mitochondrion-toxic potentials, which is why it should be avoided particularly in patients carrying POLG1 mutations or in patients with MERRF. In all patients with mitochondrial epilepsy, a ketogenic diet should be considered as a supportive measure. In some cases, a ketogenic diet may be the only effective treatment of mitochondrial epilepsy.Reference Sort, Born, Pedersen, Fonsmark and Uldall 33 Whether the application of vitamins, cofactors, or antioxidants has an additional beneficial effect on mitochondrial epilepsy has not been systematically investigated.Reference Finsterer and Zarrouk-Mahjoub 32 In single cases with MELAS syndrome, L-arginine has been shown to be beneficial not only for SLEs, but also for seizures, including status epilepticus.Reference Toribe, Tominaga, Ogawa and Suzuki 34
Headache
Headache as a feature of a MID manifests as migraine-like headache, cluster headache, nonclassified headache, or tension headache. Headache may be the dominant feature of a MID or only an ancillary feature of the phenotype. Headache may manifest as a pure manifestation of a MID or may be part of a MIMODS. For example, migraine-like headache may be an isolated manifestation of a MID or may occur together with MELAS, MERRF, chronic progressive external ophthalmoplegia (CPEO), LHON, Leigh syndrome, MIRAS, cyclic vomiting syndrome, mitochondrial depletion syndrome (MDS), or nsMIDs. Nonclassified headache has been reported in patients carrying POLG1 mutations.Reference Lam, Law and Siu 35 If headache during an SLE is resistant to L-arginine, midazolam may be effective alternatively.Reference Tsujikawa, Yokoi, Yasui, Hasegawa, Hoshiyama and Yanagi 15 Unfortunately, headache is only insufficiently described in most MID cases. Up to 58% of the patients carrying the m.3243A>G mutation develop migraine.Reference Guo, Esserlind and Andersson 36 Migraine may be also part of the clinical presentation of an SLE.Reference Wang and Le 13 The pathophysiology of migraine-like headache is poorly understood, but there are indications that it is a vascular pathology, resulting in initial hyperperfusion, which results from activation of the calcitonin-related protein or from enhanced influx of calcium into mitochondria resulting in increased oxidative stress.Reference Finsterer and Zarrouk-Mahjoub 37 Whether lactic acidosis plays a role in the development of headache in MID patients remains speculative. Only few MIDs with cluster headache have been reported.Reference Odawara, Tamaoka, Mizusawa and Yamashita 38 Treatment of headache in MIDs is the same as in non-MID patients. Migraine and migraine-like headache in MIDs may respond to nonsteroidal antirheumatic drugs, vitamin supplementation, and triptans.Reference Iizuka, Sakai, Endo and Suzuki 39 Additionally, migraine may be accessible to ketogenic diet in single patients (personal communications with patients). Headache during SLEs may respond to L-arginine or midazolam.Reference Tsujikawa, Yokoi, Yasui, Hasegawa, Hoshiyama and Yanagi 15
Ataxia
Ataxia is a frequent clinical manifestation of MIDs with CNS involvement. Ataxia in MIDs may dominate the phenotype or may be only an ancillary phenotypic feature. Ataxia may or may not be associated with a cerebellar or basal ganglia lesion. MIDs in which ataxia may dominate the phenotype include X-linked sideroblastic anemia with ataxia (XLSA), pyruvate-dehydrogenase (PDH) deficiency, NARP, MIRAS, and some nsMIDs. XLSA is characterized by early-onset sideroblastic anemia and cerebellar ataxia.Reference Allikmets, Raskind, Hutchinson, Schueck, Dean and Koeller 40 Ataxia in XLSA is usually nonprogressive, but a few cases with mild progression after the fifth decade have been reported. Ataxia predominantly manifests as gait or trunk ataxia, which may be accompanied by dysdiadochokinesia, dysmetria, dysarthria, nystagmus, hypometric saccades, strabism, or tremor.Reference Finsterer 2 Only some patients additionally develop lower-limb spasticity.Reference Hellier, Hatchwell, Duncombe, Kew and Hammans 41 Occasionally, female carriers of the X-linked forms manifest clinically.Reference Cazzola, May, Bergamaschi, Cerani, Rosti and Bishop 42 XLSA is genetically heterogeneous and may be due to mutations in the ALAS2, TRTN1, or ABCB7 genes. PDH deficiency is a rare, nonrespiratory chain associated MID resulting from mutations in the PDHA, PDHB, PDHC, and PDHD genes, which encode the four subunits of the PDH complex. PDH deficiency manifests with a wide range of abnormalities, from isolated lactic acidosis to severe Leigh syndrome.Reference Finsterer 43 Some cases may present with isolated intermittent ataxia.Reference Debray, Lambert and Gagne 44 Rarely, chromosomal defects have been reported as causative.Reference Gallant, Baldwin, Salamon, Dipple and Quintero-Rivera 45 NARP is a specific MID resulting from mutations in the ATP6 gene. It is clinically characterized by muscle weakness, ataxia, and retinitis pigmentosa. Additional phenotypic features may be learning difficulties since childhood, deafness, muscle weakness, and myoclonus. The NARP mutation m.8993T>C may also cause adult-onset myoclonus ataxia.Reference Martikainen, Gorman, Goldsmith, Burn, Turnbull and Schaefer 46 MIRAS is a mitochondrial syndrome resulting from POLG1 mutations (c.1399G>A and 2243G>C) with early-onset ataxia. Ataxia occurs as a collateral feature in MELAS, MERRF, KSS, Leigh syndrome, multiple systemic lipomatosis, MDS, sensory ataxic neuropathy, dysarthria, dilated cardiomyopathy with ataxia, pontocerebellar hypoplasia (PCH),Reference Qian, Wang, Jin, Wang, Fang, Chen and Chen 47 sensory ataxic neuropathy, dysarthria, and ophthalmoparesis, and some nsMIDs. In a study of 126 MID patients with cerebellar ataxia, 24 had pure ataxia and 102 ataxia with other MID manifestations.Reference Bargiela, Shanmugarajah and Lo 48 Among patients with idiopathic cerebellar ataxia, 28% had a MID.Reference Bargiela, Shanmugarajah and Lo 48 Ataxia in MIDs is hardly accessible to treatment, which is why only supportive measures and administration of vitamins, coenzymes, or antioxidants can be offered.
Movement Disorders
Movement disorders are a group of neurodegenerative diseases characterized by involuntary movements of the eyes, head, trunk, or limbs, at rest or during movements. Movement disorders are characterized by either paucity or excess of involuntary/asymptomatic or voluntary movements unrelated to weakness or spasticity.Reference Fahn, Jankovic and Hallett 49 Two main groups of movement disorders are delineated: the akinetic-rigid syndromes (e.g. Parkinson syndrome) and the hyperkinetic-dyskinetic syndromes (e.g. restless leg syndrome, tremor).Reference Fahn, Jankovic and Hallett 49 Any of these types of movement disorders have been occasionally described in single cases or small case series of patients with specific or nsMIDs,Reference Martikainen, Ng and Gorman 50 and there is increasing evidence that movement disorders can be a major part of the phenotypic spectrum of MIDs.Reference Finsterer 51 However, there are only a few retrospective studies commenting on movement disorders in a larger group of genetically or biochemically confirmed MIDs available. In a recent retrospective study, 42 patients with a movement disorder were identified among 678 MID patients.Reference Martikainen, Ng and Gorman 50 Almost two-thirds of the 42 cases were male. Parkinsonism was found in 13 patients and dystonia in 11. The most frequent imaging abnormality among the 42 patients was basal ganglia calcification, which was associated with generalized dystonia or Leigh syndrome.Reference Martikainen, Ng and Gorman 50 Dystonia was the most common movement disorder among pediatric patients and most commonly associated with mtDNA mutations. Parkinsonism was the most frequent movement disorder among adult MID patients and was most commonly associated with POLG1 mutations.Reference Martikainen, Ng and Gorman 50 Parkinson syndrome has been also reported in patients with a deletion of the cytb gene,Reference De Coo, Renier and Ruitenbeek 52 in MERRF,Reference Horvath, Kley, Lochmüller and Vorgerd 53 CPEO from C10orf2 mutations,Reference Kiferle, Orsucci and Mancuso 54 in nsMIDs from mutations in the STXBP1 geneReference Keogh, Daud and Pyle 55 or MPV17,Reference Garone, Rubio and Calvo 56 and in MIDs from the m.4296G>A mutation.Reference Martikainen, Kytövuori and Majamaa 57 Dystonia has been most frequently reported in MELAS, where it may be the presenting manifestation,Reference Sudarsky, Plotkin, Logigian and Johns 58 in MERRF in the form of spasmodic dysphonia,Reference Peng, Crumley and Ringman 59 in LHON,Reference Watanabe, Mita, Takita, Goto, Uchino and Imamura 60 and in nsMIDs from SUCLA2 mutationsReference Morava, Steuerwald and Carrozzo 61 or the m.8332A>G mutation.Reference Gal, Pentelenyi and Remenyi 62 Some patients with complex-I defect or PDH deficiency may develop exertion-induced dystonia.Reference Chandra and Issac 63 Paroxysmal exercise-induced dystonia may occur in patients with mitochondrial ECHS1 deficiency. Treatment of movement disorders in MIDs is not different from non-MID movement disorders, but occasionally less effective.Reference Finsterer and Bindu 25
Hypothalamic-Hypophysial-Adrenal Axis (HHAA)
Involvement of the HHAA may manifest as hypopituitarism or pituitary adenoma. Hypopituitarism may manifest as short stature, hypothyroidism, hypocorticism, hypogonadism, polydipsia, or arterial hypotonia. Hypopituitarism has been reported in MELAS,Reference Pronicki, Sykut-Cegielska and Mierzewska 64 KSS,Reference Berio and Piazzi 65 or nsMIDs from mutations in the isoleucyl t-RNA synthetase gene.Reference Schwartzentruber, Buhas and Majewski 66 Pituitary adenoma has been reported in LHONReference Mulliez, Blanckaert and Blanckaert 67 and some nsMIDs.Reference Finsterer, Stöllberger and Keller 68 Supplementation of decreased hormone levels has been tried with a beneficial effect in single cases.Reference Kang, Wang, Zhang, Pang and Gu 69
Muscle Weakness
Weakness of bulbar muscles in MIDs may occasionally be due to affection of the upper motor neuron or involvement of the intracerebral segment of the lower motor neuron. Involvement of the upper motor neuron may go along with muscle weakness and spasticity, exaggerated tendon reflexes, and positive pyramidal signs. Involvement of the intracerebral segment of the lower motor neuron can go along with muscle weakness, muscle hypotonia, and reduced tendon reflexes, such as in Leigh syndrome. There are also cases that present with spasticity but without muscle weakness and also cases with muscle hypotonia but without muscle weakness. If cranial nerves innervating bulbar muscles are affected, dysarthria, dysphagia, and tongue or facial weakness and wasting may ensue. If bulbar involvement is due to an upper motor neuron lesion, the masseter reflex may be exaggerated. Involvement of the bulbar muscles and the limb muscles together with pyramidal signs may give rise to mix up a MID with amyotrophic lateral sclerosis.Reference Finsterer and Zarrouk-Mahjoub 1 Spasticity with muscle weakness has been reported in CHCHD10 disordersReference Ait-El-Mkadem, Chaussenot, Bannwarth, Rouzier and Paquis-Flucklinger 70 and complex I deficiency.Reference Lee, Hwang, Ryu, Lee and Kim 71 Spasticity without muscle weakness has been reported in nsMIDs from an SPG7 mutation.Reference Pfeffer, Gorman and Griffin 72 Hypotonia with muscle weakness has been found in nsMIDs from PMPCA mutations.Reference Joshi, Anselm and Shi 73 Muscle hypotonia without muscle weakness has been observed in coenzyme-Q deficiencyReference Brea-Calvo, Haack and Karall 74 and other MIDs (Table 2). Only supportive measures are available to influence muscle weakness, hypotonia, and spasticity.
Psychiatric Abnormalities
The main psychiatric abnormalities associated with MIDs include cognitive deterioration including dementia, mood disorders, anxiety disorders, and psychosis.Reference Anglin, Garside, Tarnopolsky, Mazurek and Rosebush 75 More rarely reported are attention deficit hyperactivity disorder in Leigh syndrome,Reference Chuquilin, Govindarajan, Peck and Font-Montgomery 76 autism spectrum disorders,Reference Weissman, Kelley and Bauman 77 Münchausen syndrome, and bipolar disorder.Reference Munakata, Fujii, Nanko, Kunugi and Kato 78 Psychiatric disorders in MIDs may go along with or without neurological abnormalities. This is why isolated psychiatric disease has to be considered as a manifestation of a MID. Cognitive dysfunction has been occasionally reported in MIDs with diffuse cerebral lesions but not in cases with SLEs.Reference Ichikawa 3 Affected domains of cognitive function include abstract reasoning, verbal memory, visual memory, language (naming and fluency), executive or constructive functions, attention, and visuospatial function.Reference Ichikawa 3 Cognitive impairment may be a transient condition if it is due to a complex partial seizure or a permanent or even progressive condition if it is the direct manifestation of the underlying metabolic defect. Cognitive dysfunction has been reported in MELAS,Reference Ichikawa 3 MERRF, NARP,Reference Rawle and Larner 79 LHON, CPEO, KSS, mitochondrial neurogastrointestinal encephalopathy (MNGIE), Leigh syndrome, and Alpers-Huttenlocher syndrome.Reference Finsterer 80 Mitochondrial dementia has been recognized in MELAS,Reference Prasad, Narayan and Prasad 81 MERRF,Reference DiMauro and Hirano 82 KSS,Reference Phadke, Lokeshwar and Bhutada 83 CPEO,Reference Carelli, Musumeci and Caporali 84 and nsMIDs due to the m.586G>A mutation in the tRNA(Phe) gene.Reference Young, Blakely and Swalwell 85 Mood disorders, such as depression, have been observed in MELAS where it may be treatment-resistant,Reference Anglin, Garside, Tarnopolsky, Mazurek and Rosebush 75 , Reference Magner, Honzik and Tesarova 86 MERRF,Reference Molnar, Perenyi, Siska, Nemeth and Nagy 87 NARP,Reference Gelfand, Duncan and Racine 88 CPEO due to C10orf2 (twinkle) mutations,Reference Kiferle, Orsucci and Mancuso 54 , Reference Tafakhori, Yu Jin and Tohari 89 POLG1-related disorders,Reference Gurgel-Giannetti, Camargos, Cardoso, Hirano and DiMauro 90 and in nsMIDs.Reference Rapinesi, Janiri and Kotzalidis 91 An anxiety disorder as a manifestation of a MID has been described in nsMIDs.Reference Rapinesi, Janiri and Kotzalidis 91 Psychosis has been reported in MELAS,Reference Magner, Honzik and Tesarova 86 KSS,Reference Desnuelle, Pellissier, Serratrice, Pouget and Turnbull 92 POLG1-related disorders,Reference Hopkins, Somoza and Gilbert 93 infantile onset spinocerebellar ataxia,Reference Nikali and Lönnqvist 94 Leigh syndrome,Reference Mnif, Sellami and Masmoudi 95 and nsMIDs.Reference Vasconcellos, Leite, Cavalcanti, Moreira, Feijó and Souza 96 Psychiatric abnormalities particularly occur in patients with MELAS, in which 50% of cases are affected.Reference Anglin, Garside, Tarnopolsky, Mazurek and Rosebush 75 Psychiatric abnormalities in MELAS other than those described previously include borderline personality disorder,Reference Anglin, Garside, Tarnopolsky, Mazurek and Rosebush 75 confusional states,Reference Magner, Honzik and Tesarova 86 logorrhea, disinhibition, agitation, and euphoria.Reference Namer, Wolff, Dietemann and Marescaux 17 Psychiatric abnormalities may even be the presenting manifestation of MELAS.Reference Anglin, Garside, Tarnopolsky, Mazurek and Rosebush 75 Psychiatric disorders in MIDs are treated in the same way as in non-MID patients, but there are few data about mitochondrion toxicity of antipsychotic drugs available.Reference Finsterer and Bindu 25
Nystagmus
Spontaneous, gaze-evoked, or pursuit-paretic nystagmus is an infrequent clinical manifestation of a MID and rarely occurs as an isolated phenotypic feature. Together with other CNS or extra-CNS abnormalities, it has been reported most frequently in Leigh syndromeReference Kocamanoglu and Sarihasan 97 , Reference Herzer, Koch and Prokisch 98 and more rarely in LHON,Reference Nakaso, Adachi and Fusayasu 99 MELAS,Reference Choi, Kim, Oh, Jeong and Kim 100 MDS from DGUOK deficiency,Reference Ji, Dimmock and Tang 101 POLG1-related disorders,Reference Van Goethem, Luoma and Rantamäki 102 or in nsMIDs.Reference Tinsa, Ben Amor, Kaabachi, Ben Lasouad, Boussetta and Bousnina 103 - Reference Brinckmann, Rüther and Williamson 105 Downbeat nystagmus has been reported in a patient with MELAS syndrome as a result of the tRNA(Leu) mutation m.3271T>C.Reference Shinmei, Kase and Suzuki 106 Nystagmus may also be due to vestibular involvement in the MID, which can be differentiated by vestibular testing.Reference Cardenas-Robledo, Saber Tehrani, Blume and Kattah 107 Nystagmus has to be further differentiated from epileptic nystagmus.Reference Choi, Kim, Oh, Jeong and Kim 100 In a retrospective study of 59 patients with genetically confirmed MID, nine (5.3%) presented with nystagmus.Reference Grönlund, Honarvar and Andersson 108 There is no specific treatment of nystagmus available, but in some cases it may respond to nonspecific therapy with vitamins, cofactors, or antioxidants given as a general supportive treatment in MIDs.Reference Yubero, O’Callaghan and Montero 109
WMLs
WMLs are the most frequent morphological CNS abnormality of MIDs. They may or may not be accompanied by clinical manifestations, other CNS abnormalities, or non-CNS manifestations. WMLs may coexist with gray matter lesions such as in MNGIE resulting from TYMP mutations.Reference Wang, Chen and Wang 110 The morphology of WMLs in MIDs is quite variable, which is why they may be easily mixed up with other CNS disorders; other hereditary leukoencephalopathies, leukodystrophies, and multiple sclerosis particularly can be easily mixed up with WMLs in MIDs. WMLs may be categorized as spotty, patchy, confluent, centripetal or centrifugal, or as subcortical or central. MIDs with prominent white matter involvement include MELAS, MNGIE, LHON,Reference Matthews, Enzinger and Fazekas 111 KSS,Reference Ergül, Nişli, Saygılı and Dindar 112 Leigh syndrome,Reference Arii and Tanabe 113 NARP,Reference Renard and Labauge 114 PCH,Reference Lax, Alston and Schon 115 leukoencephalopathy, brainstem and spinal cord lesions, and lactic acidosis (LBSL),Reference Biancheri, Lamantea and Severino 116 and nsMIDs from a single mtDNA deletion,Reference Biancheri, Bruno, Cassandrini, Bertini, Santorelli and Rossi 117 tRNA(Trp),Reference Sanaker, Nakkestad, Downham and Bindoff 118 ECSH1,Reference Ferdinandusse, Friederich and Burlina 119 or a NDUFAF1 mutation.Reference Wu, Peng and Ma 120 In a study of 33 genetically confirmed MIDs resulting from mutations in mtDNA located genes, the SURF1, and the POLG1 gene, 18.1% had WMLs.Reference Bindu, Arvinda and Taly 121
Gray Matter Lesions
Gray matter lesions may occur as an isolated feature or together with WMLs or other cerebral abnormalities. They may be symmetric or asymmetric. They may be stable, progressive, or regressive over time.Reference Bonfante, Koenig, Adejumo, Perinjelil and Riascos 122 Most commonly, gray matter lesions occur in patients with Leigh syndrome.Reference Bonfante, Koenig, Adejumo, Perinjelil and Riascos 122 Gray matter lesions in Leigh syndrome show up as T2-hyperintensities of the caudate nucleus, putamen, tegmentum, tectum, periaqueductal area, cerebellum, or pons.Reference Bonfante, Koenig, Adejumo, Perinjelil and Riascos 122 The cortical gray matter may be involved in patients with MELAS.Reference Casimiro, Martins and Nunes 123 The periaqueductal gray matter can be affected in MERRF in addition to atrophy of the cerebellar pedunculi.Reference Ito, Shirai, Asahina and Hattori 124 Gray matter lesions together with WMLs have been described in MNGIE.Reference Wang, Chen and Wang 110
Atrophy
Atrophy may be diffuse or focal, may affect the supratentorial section or the infratentorial section, may go along with or without clinical manifestations, may be mild or severe, or may be associated with or without other CNS lesions of a MID. Cerebral atrophy occurs in specific MIDs and nsMIDs. Among the specific MIDs, atrophy is particularly prevalent in PCH,Reference Marsh, Lukic and Pope 125 CPEO,Reference Wray, Provenzale, Johns and Thulborn 126 MELAS,Reference Yokoyama, Hasegawa, Obama, Ishihara and Yagishita 127 MERRF,Reference Ito, Shirai, Asahina and Hattori 124 PDH deficiency,Reference Sofou, Steneryd, Wiklund, Tulinius and Darin 128 KSS,Reference Wray, Provenzale, Johns and Thulborn 126 and LHON.Reference Morimoto, Nagano and Deguchi 129 PCH can even show up as complete agenesis of the corpus callosum.Reference Marsh, Lukic and Pope 125 PCH is genetically heterogeneous and can be due to mutations in the AMPD2, DKC1, RARS2, PCLO, VRK1, EXOSC3, TSEN54, CASK, TSEN2, ALAAS2, ABCB7, or TET2 genes, respectively. Predominantly cortical atrophy has been reported in patients with CPEO.Reference Heidenreich, Klopstock, Schirmer, Saemann, Mueller-Felber and Auer 130 Pontine and cerebellar atrophy with a hot cross bun sign resulting from the mtDNA deletion m.3264_1607del12806 may clinically mimic the cerebellar type of multisystem atrophy (MSA-C) manifesting as dysarthria, nystagmus, falls, tremor, impaired coordination, incontinence, dysphagia, or frequent choking.Reference Alsemari and Al-Hindi 131
Basal Ganglia Calcification
Basal ganglia calcification is a rare phenotypic feature of nsMIDs and often presents without clinical manifestations and is thus often an incidental finding. Basal ganglia calcification may occur unilaterally or bilaterally and in case of bilateral occurrence it may be symmetric or asymmetric. Basal ganglia calcification may or may not be associated with other cerebral or extracerebral manifestations. Basal ganglia calcification is often attributed to non-MID causes and thus neglected as a phenotypic feature of MID. Basal ganglia calcification has been reported in specific and nsMIDs. Among the specific MIDs it has been described in MELAS,Reference Chen, Xiong, Wang, Xiong, Huang, Zhang and Wang 132 Leigh syndrome,Reference Martikainen, Ng and Gorman 50 and KSS.Reference Allen, DiMauro, Coulter, Papadimitriou and Rothenberg 133 More frequently, basal ganglia calcification can be found in nsMIDs than in specific MIDs.Reference Finsterer and Bastovansky 134 Basal ganglia calcification may even occur in pediatric patients with MELAS.Reference Liu, Bao, Ma, Chang, Qin and Wu 135 , Reference Chung, Chen, Chen and Lee 136 In single cases, basal ganglia calcification was associated with generalized dystonia.Reference Qian, Wang, Jin, Wang, Fang, Chen and Chen 47
Hypometabolism
FDG-PET reflects glucose uptake into cells. Reduced uptake into cells reflects hypometabolism within cells. In a study of five patients with Leigh syndrome, of whom four were genetically confirmed, FDG-PET showed hypometabolism in the cerebellum, the basal ganglia, and the temporal lobes.Reference Haginoya, Kaneta and Togashi 137 In one patient, hypometabolism was present despite morphologically normal cerebellum on MRI.Reference Haginoya, Kaneta and Togashi 137 In a patient with MELAS syndrome manifesting clinically as headache, seizures, and hemianopia to the right, hypometabolism on FDG-PET was demonstrated in both occipital lobes.Reference Shelly, Kelly and O’Connell 138 In two siblings with an MNGIE-like phenotype resulting from multiple mtDNA deletions, but absence of a TYMP1, POLG1, ANT1, or C10orf2 mutation, FDG-PET showed asymmetric and patchy glucose hypometabolism in the frontotemporal areas.Reference Lehnhardt, Horvath and Ullrich 139
Rare CNS Abnormalities in MIDs
Rare CNS abnormalities in MIDs include central sleep apnea syndrome, as has been described in CPEO patients,Reference Smits, Westeneng, van Hal, van Engelen and Overeem 140 and optic atrophy. Optic atrophy may be the dominant feature of a MID phenotype or a nondominant feature. As a nondominant feature, optic atrophy has been reported in dilated cardiomyopathy with ataxia syndrome.Reference Benson, Ferreira and MacDonald 141 Only in single cases was auditory agnosia reported as a CNS manifestation of an mtDNA mutation.Reference Miceli, Conti, Cianfoni, Di Giacopo, Zampetti and Servidei 142 Microcephaly may be another rare manifestation of a MID, as has been reported in an infant with MELAS syndrome.Reference Kishnani, Van Hove, Shoffner, Kaufman, Bossen and Kahler 143 Rarely, the spinal cord can be affected in nsMIDs manifesting as transverse syndrome, LBSL, or motor neuron disease.Reference Salman, Blaser, Buncic, Westall, Héon and Becker 144
Conclusions
This review shows that cerebral manifestations of MIDs can be heterogeneous and occur as isolated clinical manifestations, isolated radiologic/functional abnormalities, or as both. CNS manifestations can be the presenting feature of a MID, which is why CNS abnormalities without conclusive explanation can be indicative of a MID. If only clinical manifestations represent the onset of a MID without abnormalities on imaging or functional studies, suspecting a MID becomes difficult. The suspicion of a MID may be strengthened if there are abnormalities on imaging or functional studies in addition to the clinical manifestations. Imaging studies that strongly suggest a MID include basal ganglia calcification; symmetric gray matter lesions in the basal ganglia, brain stem, or cerebellum; or SLLs. However, the number of nonspecific findings on imaging, such as WMLs, prevail and are difficult of being attributed to a MID unless more typical manifestations in organs other than the CNS support the suspicion. The reason why certain cerebral regions are predominantly affected is unknown, but there are indications that mutation loads of maternally inherited mtDNA mutations may differ between cerebral regions and that the threshold for clinical manifestations may differ according to the local energy demand. Future studies characterizing more precisely the nature of a clinical or functional/imaging abnormality are required to improve the sensitivity of the workup. In addition to improving the diagnosis of CNS manifestations in MIDs, there is a need to improve the treatment of CNS disease in MIDs, particularly of stroke-like episodes, headache, and mitochondrial movement disorders.
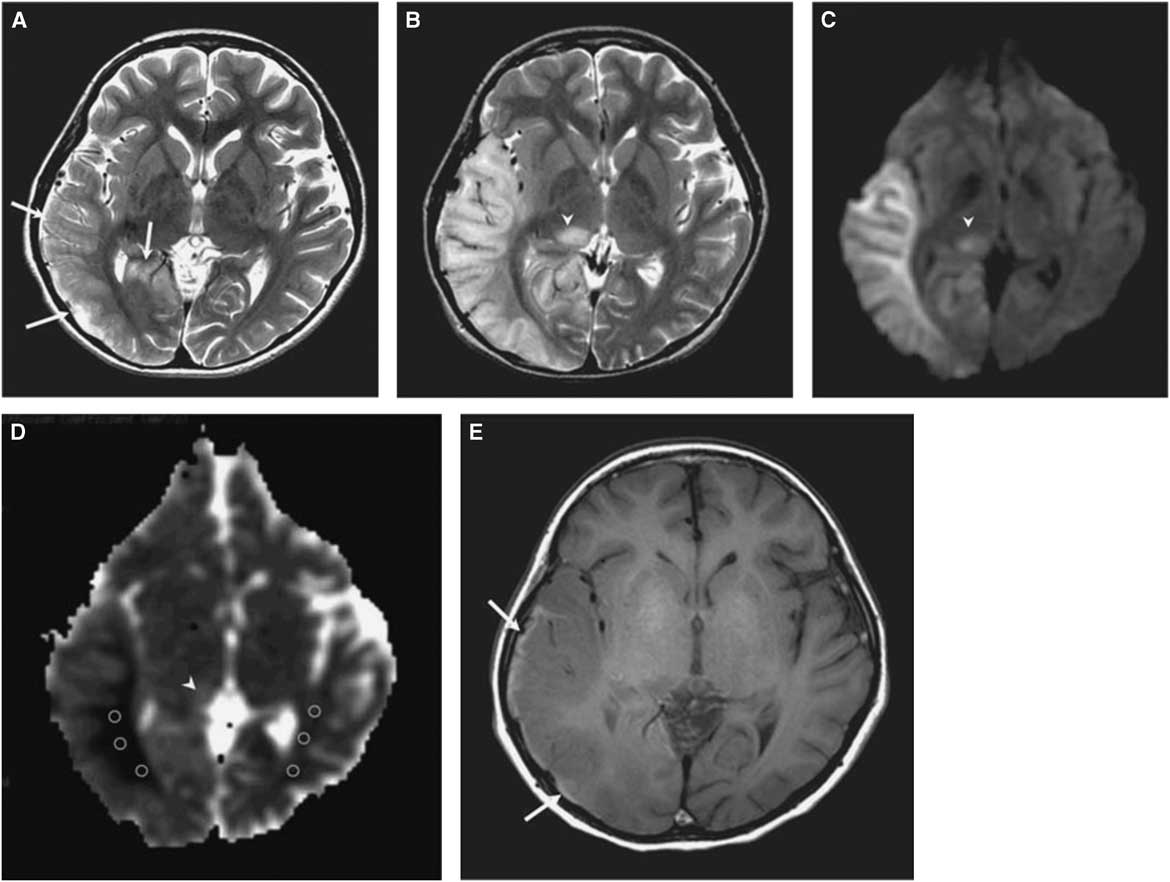
Figure 1 (A) T2-weighted image obtained at day 3 after onset of an SLE shows mild swelling (arrows) of right temporo-occipital lobe. (B) T2-weighted image obtained at day 11 after onset shows progression of edema in the right temporo-occipital lobe and newly appearing thalamic lesion (arrowhead). (C) Hyperintensity of affected areas (arrowhead) on DWI. (D) Hypointensity of the white matter and hyper-/isointensity of the cortex and thalamus on ADC (arrowhead). (E) T1-weighted image shows hyperintense rim (arrows) along cortex of swollen right temporo-occipital lobe, suggesting cortical laminar necrosis. (Reproduced from Kim et al. Korean J Radiol. 2011;12:15-24, with permission.)
Disclosures
The authors do not have anything to disclose.
Statement of Authorship
JF designed the review, organized the literature, and wrote the first draft of the manuscript. EC completed the literature search, supported in the writing, and provided critical comments.