


Vitamin A deficiency is an important public health problem in many developing nations. Particularly, marginal vitamin A deficiency is associated with increased morbidity and mortality in young children (Humphrey et al. Reference Humphrey, West and Sommer1992; Fawzi et al. Reference Fawzi, Chalmors, Herrera and Mosteller1993). Vitamin A is a generic term which describes a number of molecules exhibiting the biological activity of retinol, the precursor of naturally occurring retinoids. It is an essential nutrient required for vision, growth, embryological development, cell differentiation, reproduction, maintenance of mucous secretion and apoptosis in man and rats. The molecular mechanism of retinoic acid action mainly involves the binding and activation of specific nuclear receptors, retinoic acid receptor and retinoid X receptor, that modulate gene expression (Chambon, Reference Chambon2005). The liver is the body's main storage site for vitamin A and also regulates the secretion of retinol into the circulation in response to the demands of vitamin A-requiring target tissues (Blomhoff et al. Reference Blomhoff, Green, Berg and Norum1990).
A relationship between vitamin A nutritional status and blood lipids has been indicated. It is known that vitamin A and its derivatives cause hypertriglyceridaemia with high levels of intake (Dicken, Reference Dicken1881). On the other hand, it has been revealed that vitamin A deficiency in maturing male rats results in hypotriglyceridaemia and increased microviscosity of liver lipids (Kon'IIa et al. Reference Kon', Sokolov, Filatov, Deev and Gapparov1990) and also in a decrease of antioxidant defences leading to hepatic lipoperoxidation (Oliveros et al. Reference Oliveros, Vega, Anzulovich, Ramírez and Giménez2000; Korichneva et al. Reference Korichneva, Waka and Hammerling2003).
Acetyl-CoA carboxylase (ACC) catalyses the first step committed to fatty acid synthesis and is generally accepted to be a potentially rate-limiting enzyme in that pathway. Mammals have two major isoforms of ACC (α and β). ACCα is the major isoform in lipogenic tissues such as adipose and liver tissue, and ACCβ has a predominant location in heart and skeletal muscles (López-Casillas & Kim, Reference López-Casillas and Kim1991). The carboxylation of acetyl-CoA by ACC produces malonyl-CoA, which participates in fatty acid biosynthesis and also acts as a physiological regulator of fatty acid oxidation in the liver through inhibition of carnitine palmitoyltransferase-I (CPT-I; McGarry & Brown, Reference McGarry and Brown1997). CPT-I is an integral protein of the outer mitochondrial membrane that catalyses the initial reaction in the mitochondrial import of long-chain fatty acid-CoA where they are oxidized. This is a tightly regulated step in the cellular fatty acid utilization pathway. The enzyme exists as two isoforms encoded by separated genes, the liver-type (L-CPT-I or CPT-IA), a hepatic-enriched, ubiquitously expressed protein (McGarry, Reference McGarry1995), and the muscle-type (M-CPT-I or CPT-IB) that is highly expressed in heart, skeletal muscle, brown and white adipocytes and testes (Esser et al. Reference Esser, Brown, Cowan, Foster and McGarry1996). Retinoids have been proposed to regulate mitochondrial processes. The existence of a retinoic acid binding protein associated with mitochondria that binds and keeps retinoic acid in the mitochondria has been described (Ruff & Ong, Reference Ruff and Ong2000).
The expression of several genes involved in intra- and extracellular lipid metabolism is mediated by ligand-activated receptors, collectively referred to as PPAR. PPAR activate or repress gene expression in response to cognate ligands by binding in conjunction with the obligate heterodimerization partner, retinoid X receptor, to specific cis-acting regulatory elements called peroxisome proliferator-response elements present in the promoter regions of target genes (Berger & Moller, Reference Berger and Moller2002). Three different isoforms (α, γ and δ) have been identified and found to exhibit tissue-specific distributions; although all three exert broad regulatory effects primarily on lipid homeostasis, PPARα predominantly regulates pathways of fatty acid oxidation (Issemann et al. Reference Issemann, Prince, Tugwood and Green1993). Cross-talk between retinoid X receptor and PPARα in the liver, activated by rexinoids (selective retinoid X receptor-activators), has been reported (Ouamrane et al. Reference Ouamrane, Larrieu, Gauthier and Pineau2003).
In view of the previous observations, it was of interest to study the influence of retinol deficiency on liver fatty acid metabolism since this organ plays a major role in long-chain fatty acid and vitamin A metabolism. We examined the synthesis and oxidation of fatty acids and the mRNA expression of enzymes of regulatory importance involved in those processes. Also, the mRNA PPARα was analysed in the liver of vitamin A-deficient rats. Results were compared to the corresponding data from rats fed on an isoenergetic sufficient vitamin A diet. Finally, the effect of vitamin A restitution to vitamin A-deficient rats on lipid metabolism was analysed.
Materials and methods
Chemicals and radioisotopes
[14C]NaCO3H− (266·4 MBq/mmol), acetic acid, [1-14C]sodium salt (73·99 MBq/mmol) and [N-Me-14C]l-carnitine were purchased from Dupont, New England Company (Boston, MA, USA). Retynil-palmitate, all-trans retinol and lipids standards were acquired from Sigma Chemical Co. (St Louis, MO, USA). All other chemicals were reagent grade and were obtained from Merck Laboratory (Buenos Aires, Argentina).
Diet and experimental design
Male Wistar rats were obtained from Romanelli S.R.L. (Buenos Aires, Argentina). Rats at 21 d old were housed individually in stainless steel cages and randomly divided into two groups (eight per group) given either the experimental diet, devoid of vitamin A (vitamin A-deficient diet), or the same diet (control diet) supplemented with 4000 IU vitamin A (8 mg retinol as retinyl palmitate)/kg diet. Also, a group of eight deficient animals were fed with the control diet 15 d before being killed (vitamin A-refed group) for determination of mRNA expressions. Rats were housed in individual cages and kept in a 21–23°C controlled environment with a 12 h light–dark cycle. They were given free access to food and water throughout the entire 3 months of the experimental period. The experiment was conducted according to our committee recommendations for animal care. Diets were prepared according to AIN-93 for laboratory rodents (Reeves et al. Reference Reeves, Nielsen and Fahey1993). The diets have the following composition (g/kg): 397·5 maize starch, 100 sucrose, 132 dextrinized maize starch, 200 vitamin-free casein, 70 soyabean oil, 50 cellulose fibre, 35 AIN-93 mineral mix, 10 AIN-93 vitamin mix (devoid of vitamin A for the vitamin A-deficient diet), 3 l-cystine, 2·5 choline bitartrate and 0·014 tert-butylhydroquinone. Body weight and food intake were registered daily.
Plasma and liver retinol concentration analyses
Rats were killed by cervical dislocation at 09:00 hours. Blood samples were collected in EDTA-coated tubes. The liver was separated, immediately washed several times in ice-cold isotonic saline, blotted on paper to remove excess blood and then weighed. To minimize photoisomerization of vitamin A the plasma and tissues samples were taken under reduced yellow light and frozen in the dark at − 70°C until determination of retinol concentrations. Analyses were carried out within 1–3 weeks of obtaining the samples. Plasma and tissue retinol concentration was determined by HPLC (Bieri et al. Reference Bieri, Tolliver and Catagnani1979). Retinoids were extracted from plasma (0·5 ml) into hexane containing 5 μg butylated hydroxytoluene/ml as antioxidant for analysis. To determine tissue retinol mass, triplicate aliquots (0·2 g) of tissue were homogenized in deionized water, lyophilized and saponified in 1 ml ethanolic solution containing 0·9 mol/l potassium hydroxide for 1 h at 60°C under nitrogen atmosphere. Retinyl acetate was used as internal standard. Retinol and internal standard were extracted into hexane for analysis. Chromatography was performed on a Nucleosil 125 C-18 HPLC column with methanol–water (95:5, v/v) as the mobile phase. Retinol was detected by UV absorbance at 325 nm (Model 440; Waters Associates, Milford, MA, USA) and peak areas were calculated by integration (Spectra Physics Analytical, San Jose, CA, USA).
Serum lipid, glucose and β-hydroxybutyrate determinations
Glucose, lipid and β-hydroxybutyrate concentrations were detemined using fresh serum from rats that had been starved for 12 h. Serum glucose, cholesterol and HDL-cholesterol were determined by enzymatic methods using kits from Boehringer Mannheim Diagnostics (Indianapolis, IN, USA). Serum β-hydroxybutyrate was analysed using an enzyme colorimetric assay (Sigma Chemical Co.).
Liver acetyl-CoA carboxylase activity
The activity of ACC (EC 6.4.1.2) was measured in the cytosolic fraction of liver using [14C]NaCO3H− (266·4 MBq/mmol) according to the method of Allred & Roehrig (Reference Allred and Roehrig1978). The liver was homogenized in an Ultra Turrax T25 machine with 1·5 volumes of cold 0·3 m-manitol and centrifuged at 100 000 g for 1 h with a Beckman model L2 65B ultracentrifuge. Results were expressed in pmol [14C]NaCO3H− fixed/mg protein per min. Enzymatic assays were conducted in duplicate. Protein concentration was determined using the method of Lowry et al. (Reference Lowry, Rosebrough, Farr and Randall1951).
Incorporation of [14C]acetate into liver lipids
Liver slices (100 mg) were preincubated in 0·5 ml Krebs Ringer-glucose solution, pH 7·2, for 10 min at 37°C in a 95 % air–5 % CO2 atmosphere. Then the medium was replaced by 1·0 ml fresh Krebs Ringer solution with [14C]acetate (0·04 MBq) added and the different samples were incubated for 60 min. The reaction was stopped by addition of 0·2 ml 3 m-sulphuric acid and tissues were thoroughly washed in ice-cold Krebs Ringer solution until no more radioactivity was detected in the wash solution before storing samples at − 70°C until use. Lipids were saponified by treatment with 10 % (w/v) KOH in ethanol–water (100:15, v/v) for 3 h at 80°C. NEFA were recovered from the lower phase after acidification with 0·3 ml 1·2 m-HCl and extracted three times with petroleum ether (2·5 ml, boiling point 30–40°C). This fraction was dried down in a stream of nitrogen before counting its radioactivity. Aliquots of the non-saponified fraction (upper phase) were used for separation of the cholesterol fraction by TLC before counting its radioactivity in a Wallac 1409 DSA liquid scintillation counter. The results are expressed as μmol [14C]acetate incorporated/h per mg protein.
Incorporation of [1-14C]choline into phosphatidylcholine
Slices of liver were preincubated in 0·5 ml Krebs Ringer-glucose solution, pH 7·2, for 10 min at 37°C in a 95 % air–5 % CO2 atmosphere. After that, the medium was replaced by 0·5 ml fresh Krebs Ringer solution added with [1-14C]choline (0·04 MBq) added and the different samples were incubated for 60 min. The reaction was stopped by addition of 0·2 ml 3 m-sulphuric acid and tissues were thoroughly washed in ice-cold Krebs Ringer solution until no more radioactivity was detected in the wash solution. Phospholipid classes were separated by TLC (described earlier) and bands were scraped off and their radioactivity quantified. The results are expressed as pmol [1-14C]choline incorporated/h per mg protein.
Preparation of mitochondria
Liver mitochondria were prepared according to McGarry et al. (Reference McGarry, Mills, Long and Foster1983) by isolating them from the low-speed nuclear pellet of the original homogenate that yields mitochondria with only minimal contamination from other subcellular organelles. Briefly, livers were removed, immediately thereafter washed several times in ice-cold isotonic saline and chopped with scissors in ice-cold 0·25 mol/l sucrose. The medium was decanted and the tissue was resuspended in 10 volumes of fresh medium and then homogenized in a Teflon pestle in a 10 ml Potter-Elvehjem homogenizer mantained in ice throughout. The homogenate was centrifuged at 1000 g for 15 min. The supernatant was discarded and replaced with an equivalent volume of medium. The pellet was rehomogenized and centrifuged at 600 g for 10 min. The supernatant was used as the source for mitochondria, and it was pelleted by centrifugation at 15 000 g for 15 min. The mitochondrial pellet was washed twice with 0·25 mol/l sucrose and 0·15 m-KCl and finally resuspended in the latter and kept on ice until used (within 30 min). Protein was measured by the method of Lowry et al. (Reference Lowry, Rosebrough, Farr and Randall1951). Preparations of mitochondria were practically devoid of peroxisomes, as judged from measurements of catalase activity ( < 4 %) of the total homogenate catalase activity when assayed by the method of Chance et al. (1979).
Lipid determinations in hepatic tissue and mitochondria
The lipids from hepatic tissue and mitochondria were extracted with chloroform–methanol (2:1) according to the method of Folch et al. (Reference Folch, Lees and Sloane-Stanley1957). An aliquot of the lipid extracts was taken to determine total cholesterol, and another one to separate the different lipid fractions by TLC using plates coated with silica gel G (Merck, Darmstadt, Germany), with an n-hexane–diethyl ether–acetic acid (80:20:1, by vol.) solvent system. Lipids were detected by exposing the plates to iodine vapours. After eluting the scraped bands, aliquots were used for mass determination according to the methods of Rauser et al. (Reference Rauser, Fluster and Yamamoto1970) for phospholipids, of Sardesai & Manning (1968) for triacylglycerols and of Zak et al. (Reference Zak, Moss, Boyle and Zlatkis1954) after saponification (Abell et al. Reference Abell, Levy, Brodie and Kendall1952) for free and esterified cholesterol. On average, 93 % of cholesterol mass was recovered by TLC.
Phospholipids were separated into component species by TLC using silica gel G plates and chloroform–methanol–water (65:25:4, by vol.) as the solvent system. The individual phospholipids were identified, recovered and quantified for phosphorus content as described earlier. The results were expressed as percentage of total phospholipid phosphorus content. The position of neutral lipids and individual phospholipids was determined using the respective standard lipids.
Carnitine palmitoyltransferase-I activity
The activity of CPT-I was assayed using a method similar to that described by Grantham & Zammit (Reference Grantham and Zammit1986) with some modifications. The CPT-I activity was measured in mitochondria (400 μg protein) at 37°C in the direction: palmitoyl-CoA +[14C]carnitine → [14C]palmitoylcarnitine+CoA. Assays were performed in a medium containing 150 mmol/l KCl, 5 mm-Tris-HCl (pH 7·4), 1 mmol/l dithiothreitol, 80 μmol/l palmitoyl-CoA, 1 mmol/l EGTA, 1·3 g/l defatted bovine serum albumin, 1 mg/l rotenone and 1 mg/l antimicyn A. The mitochondria were preincubated in this medium for 5 min at 37°C before the initiation of the reaction. The reaction (500 μl final volume) was started with 0·2 mmol/l carnitine and l-[N-Me-14C]carnitine (0·04 MBq). After 4 min the reaction was stopped with 1 ml ice-cold 1·2 mol/l HCl, and water-saturated 1-butanol was added to extract the product palmitoyl-[14C]carnitine. Aliquots were assayed for radioactivity in a Wallac 1409 DSA liquid scintillation counter. Assays were run in duplicate and performed under conditions where product formation was linear with respect to both time of incubation and amount of protein.
RNA isolation and RT–PCR analysis of acetyl-CoA carboxylase, carnitine palmitoyltransferase-I and PPARα
Total RNA was isolated from 200 mg liver using TRIzol (Life Technologies, New York, USA). All RNA isolations were performed as directed by the manufacturers. Gel electrophoresis and ethidium bromide staining confirmed the purity and integrity of the samples. Quantification of RNA was based on spectrophotometric analysis at 260/280 nm. Total RNA (10 μg) was reverse-transcribed with 200 U MMLV RT (Promega Inc., Madison, WI, USA) using random hexamers as primers in a 20 μl reaction mixture, following the manufacturer's instructions. RT-generated fragments coded for β-actin (Choi & Choi, Reference Choi and Choi2000), ACC (Zhou et al. Reference Zhou, Wang, Higa, Newgard and Unger1999), CPT-I (Zhou et al. Reference Zhou, Wang, Higa, Newgard and Unger1999) and PPARα (Hoekstra et al. Reference Hoekstra, Kruijt, Van Eck and Van Berkel2003). PCR was performed in 35 μl reaction solution containing 0·2 mm-dNTP, 1·5 mm-MgCl2, 1·25 U Taq polymerase, 50 pmol of each rat specific oligonucleotide primer and RT product (1/10 of RT reaction). The sequences of the different primers are shown on Table 1. The predicted sizes of the PCR-amplified products were 243 bp for β-actin, 535 bp for ACC, 629 bp for CPT-I and 106 bp for PPARα. The samples were heated to 94°C for 2 min, followed by thirty-five temperature cycles. Each cycle consisted of three periods: (1) denaturation, 94°C for 1 min; (2) annealing, 58°C for ACC, CPT-I and β-actin, and 60°C for PPARα for 1 min; (3) extension, 72°C for 1 min. After thirty-five reaction cycles, the extension reaction was continued for another 5 min (Thermal Cycler 2400, Perkin-Elmer).
Table 1 Sequences of the primers used to amplify the different genes by RT–PCR and sizes of the fragments generated
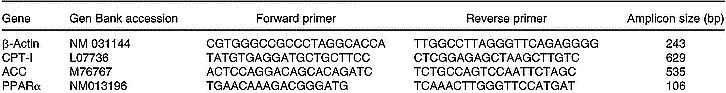
ACC, acetyl-CoA carboxylase; CPT-I, carnitine palmitoyltransferase-I.
The PCR products were electrophoresed on 2 % (w/v) agarose gel with 0·01 % (w/v) ethidium bromide. The image was visualized and photographed under UV transillumination. The intensity of each band was measured using National Institutes of Health Image software and reported as the values of band intensity units.
Statistical analyses
Data are presented as means and their standard errors. Significant differences among means were considered at a level of P < 0·05 and identified by one-way ANOVA.
Results
Body and liver weight, plasma and liver retinol, and serum lipids
The initial body weight of the rats in the three dietary groups was similar. At the time of killing, there was a significant effect of vitamin A deficiency on body weight gain, while that of the vitamin A-refed group was near to control and significantly higher than that of vitamin A-deficient rats (P < 0·05). The liver weight did not change among groups.
Since plasma retinol is not directly representative of liver vitamin A stores, the vitamin A deficiency was confirmed by analysis of liver concentrations of retinol. The plasma retinol concentrations of rats fed on the vitamin A-deficient diet were significantly lower (P < 0·001) than those of control. Total liver retinol stores of vitamin A-deficient rats were depleted, being < 7 % of the accumulated total liver retinol stores of control rats. Vitamin A refeeding considerably increased the plasma and liver vitamin A concentrations in relation to vitamin A-deficient rats.
Vitamin A deficiency resulted in a significant decrease in serum triacylglycerol, cholesterol and HDL-cholesterol concentrations, but did not modify serum VLDL + LDL-cholesterol. The serum β-hydroxybutyrate levels increased (+30 %) compared with control. In spite of a tendency toward lower serum glucose levels in the vitamin A-deficient rats compared with control rats, no statistical difference was observed. In the vitamin A-refed group serum β-hydroxybutyrate and all lipid levels reached the control values (Table 2).
Table 2 Body and liver weight, plasma and tissue retinol levels, and serum lipids and β-hydroxybutyrate in vitamin A-deficient rats (eight rats per dietary group)‡ (Mean values with their standard errors)

Mean values were significantly different from those of the control group (one-way ANOVA): *P < 0·05; **P < 0·01; ***P < 0·001.
Mean values were significantly different from those of the vitamin A-refed group (one-way ANOVA): †P < 0·05; ††P < 0·01; †††P < 0·001.
‡ For details of procedures, see pp. 264–265.
Changes in lipid profile of liver with vitamin A-deficient diet
As shown in Table 3, vitamin A deficiency affected the content of liver total phospholipids. Its concentration decreased, while cholesterol and triacylglycerol did not change in the vitamin A-deficient group in relation to those of the control group. Consequently, the ratio of total cholesterol/phospholipids increased (P < 0·05) in the liver of vitamin A-deficient rats. Vitamin A refeeding normalized the phospholipid content.
Table 3 Effect of vitamin A deficiency on liver lipid composition (μmol/g liver; eight rats per dietary group)‡ (Mean values with their standard errors)
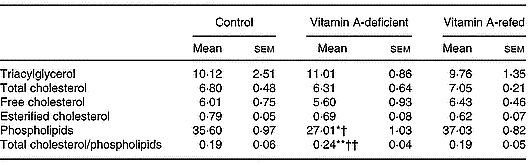
Mean values were significantly different from those of the control group (one-way ANOVA): *P < 0·05; **P < 0·01.
Mean values were significantly different from those of the vitamin A-refed group (one-way ANOVA): †P < 0·05; ††P < 0·01.
‡ For details of procedures, see pp. 264–265.
Effect of vitamin A deficiency on the synthesis of phosphatidylcholine, sphingomyelin, fatty acids and cholesterol in liver slices
The [14C]choline incorporated into phosphatidylcholine by liver was decreased by ∼43 % in the vitamin A-deficient rats, without change in sphingomyelin, when compared with control. The [14C]acetate incorporated into the saponifiable lipid fraction, indicating de novo synthesis of fatty acids, was decreased in liver from vitamin A-deficient rats (P < 0·001).
No change in non-saponifiable (cholesterol) lipid fractions was observed.
In the vitamin A-refed animals the decrease of [14C]choline incorporation into phosphatidylcholine and that of [14C]acetate into fatty acids were partially reversed (Table 4).
Table 4 Incorporation of [1-14C]acetate into saponified and non-saponified lipid fractions, and [methyl-14C]choline into phosphatidylcholine and sphingomyelin (two experiments with four rats per dietary group)‡ (Mean values with their standard errors)
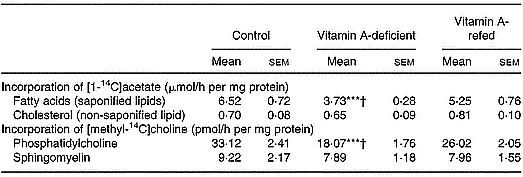
Mean values were significantly different from those of the control group (one-way ANOVA): ***P < 0·001.
Mean values were significantly different from those of the vitamin A-refed group (one-way ANOVA): †P < 0·05.
‡ For details of procedures, see pp. 264–265.
Liver acetyl-CoA carboxylase activity: effect of vitamin A deficiency
As shown in (Fig. 1), the ACC activity was significantly lower (by 36 %) in the liver of vitamin A-deficient rats than those of controls. Vitamin A refeeding in deficient animals did not completly restored the enzyme activity to control values. The cytosolic protein concentration was not modified among the three groups in the supernatant used to measure the enzyme activity (data not shown). Thus, the change observed in the enzyme activity was specifically due to change in the activity itself but it was not influenced by the total soluble protein concentration.

Fig. 1 Acetyl-CoA carboxylase (ACC) activity in the liver of control, vitamin A-deficient and vitamin A-refed rats. For details of procedures, see pp. 264–265. Values are means with their standard errors depicted by vertical bars (eight rats per dietary group). Mean value was significantly different from that of the control group: *P < 0·001. Mean value was significantly different from that of the vitamin A-refed group: †P < 0·01.
Mitochondrial carnitine palmitoyltransferase-I activity: effect of vitamin A deficiency
The CPT-I activity in the liver mitochondria of rats fed on the vitamin A-deficient diet was increased by 40 % of the control value. This increase was reverted by vitamin A refeeding, reaching values similar to control (Fig. 2).

Fig. 2 Carnitine palmitoyltransferase-I (CPT-I) activity in the liver mitochondria of control, vitamin A-deficient and vitamin A-refed rats. For details of procedures, see pp. 264–265. Values are means with their standard errors depicted by vertical bars (eight rats per dietary group). Mean value was significantly different from those of the control group and the vitamin A-refed group: *P < 0·001.
Liver mitochondria lipid content
The content of the different mitochondrial lipids of liver is showed in Table 5. The concentration of triacylglycerols increased and total phospholipid contents decreased in the vitamin A-deficient group in relation to that of the control group. The vitamin A refeeding partially restored the phospholipid content. In spite of a tendency toward recovering the triacylglycerol mass and cholesterol/phospholipid ratio after vitamin A refeeding, no statistical differences were observed compared with vitamin A-deficient rats. No changes in cholesterol contents among the three groups were observed.
Table 5 Effect of vitamin A deficiency on liver mitochondrial lipid compositions (mg/g protein; eight rats per dietary group)‡ (Mean values with their standard errors)
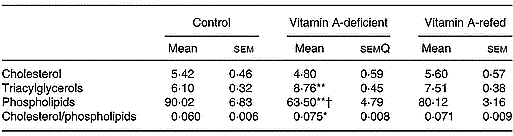
Mean values were significantly different from those of the control group (one-way ANOVA): *P < 0·05; **P < 0·01.
Mean values were significantly different from those of the vitamin A-refed group (one-way ANOVA): †P < 0·05.
‡ For details of procedures, see pp. 264–265.
On a percentage basis the phospholipid composition was modified in the mitochondria of vitamin A-deficient rats, showing a significant decrease in cardiolipin content (P < 0·01) compared with controls. No change in phosphatidylcholine, phosphatidylethanolamine, sphingomyelin and phosphatidylinositol + phosphatidylserine content was observed (Table 6).
Table 6 Effect of the diet on phospholipid compositions of liver mitochondria (percentage of total lipid phosphorus (w/w); eight rats per dietary group)‡ (Mean values with their standard errors)
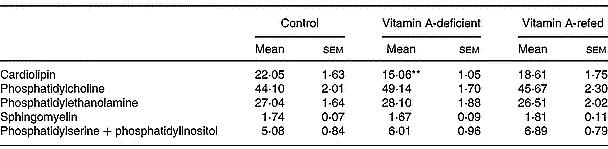
Mean values were significantly different from those of the control group (one-way ANOVA): **P < 0·01.
‡ For details of procedures, see pp. 264–265.
Vitamin A deficiency on the levels of mRNA expression of carnitine palmitoyltransferase-I, acetyl-CoA carboxylase and PPARα
As shown in Fig. 3, the expression of CPT-I mRNA, the main regulator of fatty acid β-oxidation, was increased in vitamin A-deficient rats (P < 0·01) as compared to controls and totally restored by the vitamin A refeeding. The expression of ACC mRNA was significantly lower in liver from vitamin A-deficient rats than those from control and vitamin A-refed groups. The restitution of vitamin A to the diet of vitamin A-deficient rats did not completely restore the ACC mRNA expression to control values.

Fig. 3 Expression of genes involved in liver fatty acid metabolism. For details of procedures, see pp. 264–265. Bars show quantification of the intensity of the fragment bands in relation to the intensity of the internal control bands. Values are means with their standard errors depicted by vertical bars (four rats per dietary group). +A, control; − A, vitamin A-deficient; R, vitamin A-refed rats. Mean value was significantly different from that of the +A group: *P < 0·001; **P < 0·01. Mean value was significantly different from that of the R group: †P < 0·01. The photographs are ethidium bromide-stained agarose gels of carnitine palmitoyltransferase-I PCR products (A); acetyl-CoA carboxylase PCR products (B); PPARα PCR products (C); β-actin PCR products, used as an internal control (D). The results are typical of four independent observations.
A significant decrease of PPARα mRNA expression (P < 0·001) was observed in vitamin A-deficient rats in relation to control. The vitamin A refeeding reverted that change, reaching values similar to control indicating that PPARα could be involved in the effects of vitamin A deficiency.
Discussion
The present in vivo study was undertaken to investigate the effect of vitamin A deprivation on liver fatty acid metabolism. The quantity of retinol stored in the neonatal liver during the nursing period is greatly influenced by maternal diet during lactation. We did not restrict the vitamin A intake of the dams of the experimental rats, and for this reason we prolonged the vitamin A dietary deficiency for 3 months. Plasma retinol levels alone do not reliably predict liver stores unless the plasma level reaches < 0·35 μmol/l (Olson, Reference Olson1982). In the present study, the 3 months of vitamin A deprivation after weaning produced subclinical plasma retinol concentration and negligible total retinol stores in the liver; the data confirmed vitamin A deficiency. Lower body weights of vitamin A-depleted rats has also been shown by other authors (Wright et al. Reference Wright, Wang, Fultz, Arif, Matthews and Chertow2002). We have previously demonstrated that deprivation of vitamin A for 3 months does not affect daily food intake in relation to control rats (Oliveros et al. Reference Oliveros, Vega, Anzulovich, Ramírez and Giménez2000).
It is well established that liver phospholipid content decreases in rats with vitamin A deficiency (Khanna & Reddy, Reference Khanna and Reddy1983) and, by contrast, it increases in guinea pigs with the administration of 30 mg retinol for 7 d (Alarcon Corredor et al. Reference Alarcon Corredor, Silva Larralte, Chacon Patino, Pachano Primera, Alarcon and Reinosa1996). In the present study, we have also observed a lower content of total phospholipids in liver of vitamin A-deficient rats. This was associated with a lower synthesis of the major phospholipid since [14C]choline incorporation into phosphatidylcholine decreased. In addition, the decreased incorporation of [14C]acetate into saponifiable lipids and the low activity of ACC indicated a decrease in fatty acid synthesis, suggesting a low availability of fatty acids to form phospholipids in the liver of vitamin A-deficient rats. These metabolic changes can possibly affect the serum lipid levels. In particular, phospholipids are an important component of HDL, the level of which is reduced in serum of vitamin A-deficient rats.
Vitamin A deficiency did not alter liver cholesterol synthesis, since incorporation of [14C]acetate into cholesterol was not modified compared with control. However, there was a decrease of the serum cholesterol associated with a low HDL-cholesterol level. Although the mechanism of this hypolipidaemic effect is not the purpose of the present study, the possibility that vitamin A deficiency may play a role in altering lipoprotein secretion from the liver into the circulation can be considered. It has been shown that retinoic acid increases in a dose-dependent manner the secretion of apo A I, B 100, C III and A II in cultures of Hep G2 cells (Liu et al. Reference Liu, Wu and Liu2001). In addition, we have observed a decrease in serum triacylglycerol levels in vitamin A-deficient rats.
Fatty acid utilization through the mitochondrial β-oxidation pathway was clearly affected by the vitamin A deficiency. The increase in serum β-hydroxybutyrate was attributed to high CPT-I activity in liver mitochondria of vitamin A-deficient rats. This fact was associated with the low ACC activity, which catalyses the formation of malonyl-CoA (López-Casillas & Kim, Reference López-Casillas and Kim1991), a strong inhibitor of synthesis and activation of CPT-I (Jackson et al. Reference Jackson, Zammit and Price2000). Malonyl-CoA sensitivity is an intrinsic property of L-CPT-I since carnitine palmitoyltransferase-II does not possess a malonyl-CoA binding domain (Cohen et al. Reference Cohen, Kohl, McGarry, Girard and Prip-Buus1998).
Furthermore, the high CPT-I activity coincided with an enhanced CPT-I mRNA level, and the low ACC activity was in agreement with a decreased ACC gene. The incorporation of vitamin A into the diet of vitamin A-deficient rats considerably improved both activities and genes expressions of CPT-I and ACC.
To gain more insight into the control of fatty acid metabolism by vitamin A deficiency, we have examined the transcriptional regulation of the PPARα gene, which plays a pivotal role in the transcriptional regulation of genes involved in cellular lipid metabolism (Issemann et al. Reference Issemann, Prince, Tugwood and Green1993; Kersten, 2002). The low PPARα mRNA expression in vitamin A-deficient rats could be explained by low fatty acid synthesis and the depletion of retinoids, since fatty acids are able to activate PPARα (Forman et al. Reference Forman, Chen and Evans1997), and 9-cis retinoic acid is the ligand for the retinoid X receptor, which in turn acts as the dimerization partner for PPARα. The expression of the PPARα gene was restored with a vitamin A-sufficient diet, indicating that it is involved in the effects of vitamin A deficiency on liver fatty acid metabolism.
We also observed a significant decrease of total phospholipid content in mitochondria of vitamin A-deficient rats given an increased cholesterol–phospholipid relation, suggesting that membrane fluidity can be altered. Kon'IIa et al. (Reference Kon', Sokolov, Filatov, Deev and Gapparov1990) found that vitamin A deficiency in maturing male rats changes the lipid composition of liver microsomal membranes increasing the cholesterol/phospholipid ratio. We have seen a significant decrease (by 36 %) of mitochondrial cardiolipin content induced by vitamin A deficiency that could also indicate a mitochondrial dysfunction. Cardiolipin is recognized to be an essential phospholipid in eukaryotic energy metabolism and in maintaining mitochondrial structure and function. A direct relationship between cardiolipin loss and cytochrome c release from mitochondria has been identified as an initial step in the phatway to apoptosis (McMillin & Dowhan, Reference McMillin and Dowhan2002). In addition, in a previous report we showed that vitamin A deprivation for 3 months induces oxidative stress in rat liver (Anzulovich et al. Reference Anzulovich, Oliveros, Muñoz, Martínez and Gimenez2000). It is known that reactive oxygen species affect the activity of heart mitochondrial complex III leading to mitochondrial dysfunction via cardiolipin oxidative damage. This has been mainly ascribed to a specific loss in mitochondrial content of cardiolipin (Petrosillo et al. Reference Petrosillo, Ruggiero, DiVenosa and Paradies2003). The increased triacylglycerol content could be due to a decrease in the lipid membrane turnover, as has been suggested by Barber et al. (Reference Barber, Borrás, Torres, García, Cabezuelo, Lloret, Pallardó and Viña2000), who found changes in lipid composition of liver mitochondria associated with oxidative damage induced by chronic vitamin A deficiency.
Considering all these observations, it is also conceivable that modified mitochondria lipid composition can alter the input of fatty acids and consequently their β-oxidation in the liver of vitamin A-deficient rats.
The present results confirm and extend the observation that vitamin A depletion rendered important alterations in total liver lipid metabolism and mitochondrial lipid compositions. In particular, the results show that 3 months of feeding the vitamin A-deficient diet to the rats causes a significant increase in mitochondrial fatty acid oxidation by an enhancement of CPT-I activity and gene expression. This is mainly attributed to a decrease in availability of malonyl-CoA due to a diminution of ACC activity and gene expression. The present observation establishes an explanation for vitamin A action, which could control energetic mitochondrial processes in situations such as retinoic treatment.
Acknowledgements
María Sofía Gimenez is a member of the National Investigations Council of Science and Technology (CONICET), Argentina.









