


Numerous epidemiological and animal studies demonstrate a link between poor in utero environment and the development of cardiovascular and metabolic abnormalities in the offspring(Reference McMillen and Robinson1). There is growing evidence that environmental challenges during pregnancy in the F0 generation can induce metabolic effects not only in the F1 generation but also in the F2 generation(Reference Zamenhof, Van Marthens and Grauel2–Reference Burdge, Slater-Jefferies, Torrens, Phillips, Hanson and Lillycrop8).
We have previously reported that feeding a maternal protein-restricted diet during pregnancy leads to raised blood pressure and endothelial dysfunction in the F1 offspring(Reference Brawley, Itoh, Torrens, Barker, Bertram, Poston and Hanson9, Reference Torrens, Brawley, Anthony, Dance, Dunn, Jackson, Poston and Hanson10), which persists into pregnancy in the F1 females(Reference Torrens, Brawley, Barker, Itoh, Poston and Hanson11). This may be clinically relevant to humans because the vascular endothelium plays a major role in the control of vascular tone, through the release of factors such as NO, prostacyclin and endothelial-derived hyperpolarising factor(Reference Busse and Fleming12), while endothelial dysfunction is associated with both atherosclerosis(Reference Landmesser, Hornig and Drexler13) and hypertension(Reference Taddei, Virdis, Ghiadoni, Sudano and Salvetti14). Vascular endothelial dysfunction is also recognised as a common feature of the metabolic syndrome(Reference Fornoni and Raij15).
Sophisticated genomic analysis is now being used to determine possible genes associated with increased risk of myocardial infarction, type 2 diabetes and hypertension(Reference Broeckel, Hengstenberg and Mayer16, Reference Zeggini, Weedon and Lindgren17). It is also well-established that a substantial proportion of heritable risk is transmitted through non-genomic mechanisms(Reference Gluckman, Hanson and Beedle18). Some candidate gene pathways involved in metabolism have been demonstrated to be modified by epigenetic processes in both F1 and F2 generations(Reference Burdge, Slater-Jefferies, Torrens, Phillips, Hanson and Lillycrop8). Effects on cardiovascular function have, however, not been shown. To test whether blood pressure and vascular dysfunction, induced by maternal dietary imbalance during pregnancy, could be passed to the F2 generation we assessed blood pressure and small mesenteric artery function in the F2 offspring of protein-restricted rat dams.
Methods
All animal procedures were in accordance with the regulations of the British Home Office Animals (Scientific Procedures) Act, 1986 and the present study was approved by the local ethical review committee.
Animals and dietary protein restriction
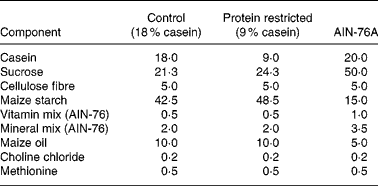
Virgin female Wistar rats were mated (the day of vaginal plug detection being defined as day 0 of pregnancy) and pregnant rats were randomly assigned to two dietary groups: control (18 % casein, n 9) or an isoenergetic protein-restricted diet (PR, 9 % casein, n 7; see Table 1). Pregnant dams were fed on the experimental diets from confirmation of pregnancy until delivery as previously described(Reference Torrens, Brawley, Barker, Itoh, Poston and Hanson11). Immediately postpartum, dams and F1 pups were returned to standard laboratory chow (AIN-76A; Table 1) and continued receiving standard chow for the remainder of the experiment. At age 124 (sem 10) d, one female offspring from each litter was randomly selected and mated (control n 9; PR n 7) with breeding males from outside the colony, and which were not subject to dietary manipulations, to produce an F2 offspring of female F1 lineage. The same males were available to both control and PR groups to prevent a possible bias arising from the breeding males.
Table 1 Composition of experimental diets (g/100 g)
Blood pressure measurement
To coincide with F1 offspring(Reference Brawley, Itoh, Torrens, Barker, Bertram, Poston and Hanson9), systolic blood pressure was recorded at approximately age 100 d in F2 offspring by tail-cuff plethysmography using an IITC blood pressure monitor (229 model; Linton Instruments, Diss, Norfolk, UK)(Reference Brawley, Itoh, Torrens, Barker, Bertram, Poston and Hanson9). Briefly, animals whose dietary group was blinded to the investigator were placed in a room heated to 29°C to stimulate tail blood flow. Once acclimatised to the temperature, rats were placed in a restraint tube and an appropriate-sized cuff placed over the tail and inflated. Blood pressures were determined in triplicate for each animal and the mean recorded. To minimise the stressful response, animals were handled by trained staff throughout their life and were made familiar with the recording equipment before measurements being made.
Vascular protocol
Male F2 offspring were randomly selected from each litter and killed at both 80 and 200 d by CO2 inhalation and cervical dislocation. Small mesenteric arteries (internal diameter about 300 μm and known to contribute to peripheral resistance(Reference Christensen and Mulvany19)) were dissected, cleaned of connective tissue and mounted in a wire myograph (Danish Myo Technology A/S, Aarhus, Denmark). Segments were bathed in physiological salt solution (PSS) (NaCl, 119 mm; KCl, 4·7 mm; CaCl2, 2·5 mm; MgSO4, 1·17 mm; NaHCO3, 25 mm; KH2PO4, 1·18 mm; EDTA, 0·026 mm; d-glucose, 5·5 mm), heated to 37°C and continuously gassed with 95 % O2 and 5 % CO2. The passive tension–internal circumference relationship (IC100) was determined by incremental increases in tension to achieve an internal circumference equivalent to a transmural pressure of 100 mmHg (using the Laplace relationship) and the arteries were set to a diameter equivalent to 0·9 × IC100 as previously described(Reference Mulvany and Halpern20). Functional integrity of the smooth muscle was assessed with four 2 min washes with 125 mm-KPSS solution (PSS with an equimolar substitution of KCl for NaCl). Vessels failing to produce an active tension equivalent to 13·3 kPa were discarded from the study.
At the day 80 time point, cumulative concentration response curves to the α1-adrenoceptor agonist phenylephrine (PE; 10 nm to 100 μm) and the thromboxane mimetic U46619 (10 pm to 1 μm) were conducted. Vessels were preconstricted with a submaximal dose of PE equivalent to EC80 (effective concentration equal to 80 % of maximal concentration) and concentration response curves to the endothelium-dependent vasodilators acetylcholine (ACh; 1 nm to 10 μm) and bradykinin (1 pm to 10 μm) and the NO donor sodium nitroprusside (100 pm to 30 μm) were performed.
At 200 d response curves to PE and ACh were repeated as at the earlier time point. At this time point the different components (NO, prostacyclin and endothelial-derived hyperpolarising factor) of ACh-mediated vasodilatation were investigated by repeated responses to ACh in the presence of the NO synthase inhibitor N ω-nitro-l-arginine methyl ester (l-NAME; 100 μm) alone, in combination with the cyclo-oxygenase inhibitor, indomethacin (INDO; 10 μm) or in combination with INDO (10 μm) and K+(25 mm) to determine the relative contributions of NO, prostacyclin and endothelial-derived hyperpolarising factor respectively to the ACh-induced vasodilatation. All chemicals were obtained from Sigma (Poole, Dorset, UK).
Calculations and statistical analysis
All data are expressed as mean values with their standard errors. Constrictor responses were calculated as percentage maximum contraction induced by 125 mm-KPSS and relaxant responses as percentage reversal of PE-induced contraction. Cumulative concentration response curves to agonists were analysed by fitting to a four-parameter logistic equation using non-linear regression to obtain the effective concentration equal to 50 % of maximum (pEC50) and a maximal response, which were compared by one-way ANOVA (Prism 3.0; GraphPad Software Inc., San Diego, CA, USA). Significance was accepted if P < 0·05. At all points the investigator was blinded to the dietary group.
Results
Growth and development
The growth of the F1 female offspring has been previously described by Torrens et al. (Reference Torrens, Brawley, Barker, Itoh, Poston and Hanson11). At time of mating there was no difference in age or starting weights of the female F1 offspring (age: control 125 (sem 10) d, n 9; PR 123 (sem 13) d, n 7; NS; weight: control 203·9 (sem 4·1) g, n 9; PR 198·1 (sem 5·9) g, n 7; NS). In the F2 offspring there was no difference in litter size or birth weights between the two groups (controls 6·11 (sem 0·20) g, n 9; PR 7·07 (sem 0·72) g, n 7; NS).
Offspring growth was similar between the groups and body weight at post-mortem was similar at both 80 d (control 278·6 (sem 8·9) g, n 14; PR 298·7 (sem 9·8) g, n 15; NS) and 200 d (control 417·0 (sem 24·5) g, n 10; PR 377·4 (sem 20·8) g, n 8; NS).
Blood pressure
Compared with controls, systolic blood pressure was raised at age 100 d in both male (control 122·1 (sem 2·3) mmHg, n 7; PR 134·7 (sem 3·2) mmHg, n 6; P < 0·01) and female (control 117·9 (sem 2·2) mmHg, n 6; PR 129·3 (sem 2·4) mmHg, n 6; P < 0·01) PR F2 offspring (Fig. 1 (a)).

Fig. 1 (a) Systolic blood pressure in age 100 d male and female F2 offspring from control (C, □, n 6–7) or protein-restricted (PR, ■, n 6) dams and (b) cumulative additions of the α1-adrenoceptor agonist phenylephrine (PE) to isolated mesenteric arteries of age 80 d F2 offspring of control (○, n 13) or PR (●, n 15) dams. Values are means, with their standard errors represented by vertical bars. ** Mean value was significantly different from that of the control group (P < 0·01).
Vascular reactivity – day 80
The internal diameter of the arterial segments mounted was similar between the groups (control 299 (sem 5) μm, n 48; PR 286 (sem 7) μm, n 35; NS).
Vasoconstriction
At 80 d, the constriction produced in response to 125 mm-KPSS was similar between the groups (controls 18·96 (sem 0·62) kPa, n 13; PR 18·63 (sem 0·61) kPa, n 15; NS). Cumulative additions of the α1-adrenoceptor agonist PE and the thromboxane mimetic U46619 both produced concentration-dependent constriction in all arteries. In F2 PR offspring sensitivity to PE was enhanced compared with controls (pEC50:PE control 5·88 (sem 0·01), n 13; PR 6·07 (sem 0·01), n 15; P < 0·001; Fig. 1 (b)) with no change in the maximal response. Vasoconstriction to U46619 tended to be increased in the PR group, but did not reach significance (pEC50:control 7·33 (sem 0·06), n 13; PR 7·45 (sem 0·05), n 11; NS).
Endothelial-dependent vasodilatation
Preconstriction to PE did not differ between the two groups (data not shown). The endothelial-dependent vasodilator ACh produced a concentration-dependent vasodilatation of PE-induced tone in all mesenteric arteries at both time points. In the PR group the maximal response to ACh was greatly attenuated compared with controls (% maximal response: control 89·7 (sem 2·6), n 14; PR 72·7 (sem 4·4), n 15; P < 0·01; Fig. 2 (a)). The endothelial-dependent vasodilator bradykinin produced a concentration-dependent vasodilatation of PE-induced tone in all arteries, which was attenuated at concentrations greater than 100 nm but did not differ between the two groups (Fig. 2 (b)).

Fig. 2 Cumulative additions of (a) the endothelium-dependent vasodilator acetylcholine (ACh), (b) the endothelium-dependent vasodilator bradykinin and (c) the endothelium-independent NO donor sodium nitroprusside (SNP) to isolated mesenteric arteries of age 80 d F2 offspring of control (○, n 13–14) or protein-restricted (●, n 9–15) dams. Values are means, with their standard errors represented by vertical bars. Mean value of percentage maximal response was significantly different from that of the control group: * P < 0·05, ** P < 0·01.
Endothelial-independent vasodilatation
Preconstriction to PE did not differ between the two groups (data not shown). The NO donor sodium nitroprusside produced a concentration-dependent vasodilatation of PE-induced tone in both groups which was enhanced in the PR group compared with controls (% maximal response: control 77·3 (3·3), n 14; PR 87·5 (2·4), n 9; P < 0·05; Fig. 2 (c)).
Vascular reactivity – day 200
The internal diameter of mounted arterial segments at the 200 d time point did not differ between the two groups (control 304 (7) μm, n 21; PR 303 (11) μm, n 15; NS).
Vasoconstriction
As at the earlier time point, at age 200 d the constriction produced in response to 125 mm-KPSS was similar between the groups (controls 18·86 (sem 0·83) kPa, n 10; PR 17·62 (sem 1·12) kPa, n 8; NS). Whilst vasoconstriction to the PE was still enhanced in arteries from the PR group compared with controls, this was seen as an increase in maximal constriction rather than enhanced sensitivity (control 102·7 (sem 1·0) % maximal response, n 10; PR 109·4 (sem 0·6) % maximal response, n 8; P < 0·05).
Endothelial-dependent vasodilatation
As at the earlier time point, preconstriction to PE did not differ between the two groups (data not shown) and impaired endothelium-dependent vasodilatation to ACh was again observed in the PR group, but this was now seen as a decrease in sensitivity (pEC50:control 7·67 (sem 0·10), n 10; PR 7·28 (sem 0·07), n 8; P < 0·05; Fig. 3 (a)).

Fig. 3 Cumulative additions of the endothelium-dependent vasodilator acetylcholine (ACh) to isolated mesenteric arteries of male age 200 d F2 offspring of (a) control (○, n 10) or protein-restricted (●, n 8) dams. Values are means, with their standard errors represented by vertical bars. * Mean value of effective concentration equal to 50 % of maximum (pEC50) was significantly different from that of the control group (P < 0·05). (b) Cumulative additions of ACh to isolated mesenteric arteries of male age 200 d control F2 offspring alone (○, n 10) or in the presence of N ω-nitro-l-arginine methyl ester (l-NAME; 100 μm, ●, n 6), l-NAME (100 μm) and indomethacin (INDO) (10 μm, ■, n 5) or l-NAME (100 μm), INDO (10 μm) and K+(25 mm, ▲, n 3). ** Mean value of percentage maximal response for the l-NAME preparations was significantly different from that of the naive preparations (P < 0·01). †† Mean value of pEC50 for the l-NAME/INDO preparations was significantly different from that of the naive preparations (P < 0·01). ‡‡‡ Mean value of percentage maximal response for the l-NAME/INDO/K+ preparations was significantly different from that of the naive preparations (P < 0·001). (c) Cumulative additions of ACh to isolated mesenteric arteries of male age 200 d protein-restricted F2 offspring alone (○, n 8) or in the presence of l-NAME (100 μm, ●, n 6), l-NAME (100 μm) and INDO (10 μm, ■, n 5) and l-NAME (100 μm), INDO (10 μm) and K+(25 mm, ▲, n 5). ‡‡‡ Mean value of percentage maximal response for the l-NAME/INDO/K+ preparations was significantly different from that of the naive preparations (P < 0·001).
Nitric oxide, prostacyclin and endothelial-derived hyperpolarising factor inhibition
In control F2 offspring, l-NAME (100 μm) significantly blunted the ACh-induced vasodilatation which was most profound at doses above 100 nm-ACh and led to a significant blunting of the maximal response (% maximal response; naive 80·2 (sem 3·2), n 10; inhibitor 54·9 (sem 8·9), n 6; P < 0·01 v. naive preparation). The combination of l-NAME with the cyclo-oxygenase inhibitor INDO (10 μm) not only attenuated the maximal response as above, but led to a significant right-ward shift in the ACh response (pEC50:naive 7·67 (sem 0·10), n 10; inhibitors 6·88 (sem 0·28), n 5; P < 0·01 v. naive preparations; Fig. 3 (b)). The presence of a depolarising K+ solution (25 mm) in addition to the l-NAME and INDO abolished any relaxation to ACh (% maximal response: naive 80·2 (sem 3·2), n 10; inhibitors − 11·5 (sem 2·3), n 3; P < 0·001; Fig. 3 (b)).
In the protein- restricted F2 offspring, incubation with l-NAME (100 μm) alone had little effect on ACh-mediated vasodilatation (% maximal response: naive 85·0 (sem 3·2), n 8; inhibitor 76·9 (sem 6·9), n 6; NS). Incubation with both INDO (10 μm) and l-NAME similarly failed to produce a significant shift in the ACh response (pEC50:naive 7·28 (sem 0·07), n 8; inhibitors 6·85 (sem 0·67), n 5; NS). However, as in the control group a depolarising K+ solution (25 mm) in addition to the l-NAME and INDO totally abolished any relaxation to ACh (% maximal response; naive 85·0 (sem 3·2), n 6; inhibitors − 11·3 (sem 3·6), n 3; P < 0·0001; Fig. 3 (c)).
Discussion
Classical risk factors for cardiovascular disease include postnatal environmental factors such as smoking, diet, physical activity as well as genetic predisposition. Recent evidence, however, highlights the importance of the in utero environment in the development of cardiovascular and metabolic disease(Reference McMillen and Robinson1). Previously, we and others have demonstrated in the rat that maternal protein restriction in pregnancy leads to raised systolic blood pressure and endothelial dysfunction in the offspring(Reference Brawley, Itoh, Torrens, Barker, Bertram, Poston and Hanson9, Reference Torrens, Brawley, Anthony, Dance, Dunn, Jackson, Poston and Hanson10, Reference Langley and Jackson21). In the present study we now demonstrate that both raised systolic blood pressure and endothelial dysfunction can be transmitted to the F2 offspring in the absence of additional dietary challenges in the F1 female offspring, either after weaning or during pregnancy.
The effect of maternal protein restriction in pregnancy on blood pressure in the offspring has been consistently demonstrated(Reference Langley and Jackson21). Previously, we have reported blood pressure increases in the order of about 20 mmHg in the F1 offspring(Reference Brawley, Itoh, Torrens, Barker, Bertram, Poston and Hanson9, Reference Torrens, Brawley, Anthony, Dance, Dunn, Jackson, Poston and Hanson10), which is comparable with the effect observed in the present study in both the male and female F2 offspring. Raised blood pressure has been observed in a number of models of development programming (for a review, see McMillen & Robinson(Reference McMillen and Robinson1)), yet to date this has not been shown to be transgenerational. More recently, a number of these models have demonstrated that the raised blood pressure is associated with vascular and in particular endothelial dysfunction(Reference Brawley, Itoh, Torrens, Barker, Bertram, Poston and Hanson9, Reference Torrens, Brawley, Anthony, Dance, Dunn, Jackson, Poston and Hanson10, Reference Holemans, Gerber, Meurrens, De Clerck, Poston and Van Assche22–Reference Armitage, Lakasing, Taylor, Balachandran, Jensen, Dekou, Ashton, Nyengaard and Poston25), a condition also associated with metabolic and cardiovascular disease in humans(Reference Landmesser, Hornig and Drexler13, Reference Taddei, Virdis, Ghiadoni, Sudano and Salvetti14, Reference Rizzoni, Porteri and Guelfi26). The finding of endothelial dysfunction alongside the raised blood pressure could suggest a role for impaired vascular control in blood pressure regulation, at least in the male F2 offspring(Reference Taddei, Virdis, Ghiadoni, Sudano and Salvetti14). A similar situation may also be occurring in the females, but unfortunately there were insufficient viable female arterial segments from the current cohort to assess this. Interestingly, although the findings of raised blood pressure and endothelial dysfunction in the present study parallel what has been previously reported in the F1 offspring of protein-restricted dams(Reference Brawley, Itoh, Torrens, Barker, Bertram, Poston and Hanson9, Reference Torrens, Brawley, Anthony, Dance, Dunn, Jackson, Poston and Hanson10), this does not appear to be the case for the maternal high-fat model where transmission to the F2 was not observed(Reference Armitage, Ishibashi, Balachandran, Jensen, Poston and Taylor27).
Evidence from the F1 offspring indicates a dysfunction in the NO/cGMP pathway(Reference Brawley, Itoh, Torrens, Barker, Bertram, Poston and Hanson9, Reference Torrens, Brawley, Anthony, Dance, Dunn, Jackson, Poston and Hanson10), which has also been implicated in the offspring of globally restricted dams through increased oxidative stress(Reference Franco, Fortes and Akamine23, Reference Franco, Arruda, Dantas, Kawamoto, Fortes, Scavone, Carvalho, Tostes and Nigro28). NO is an important vascular mediator(Reference Busse and Fleming12) and its importance is highlighted in the hypertension and vascular dysfunction seen in endothelial nitric oxide synthase (eNOS) knock-out mice(Reference Waldron, Ding, Lovren, Kubes and Triggle29). From the present study the role for dysfunction in the NO pathway is unclear. The attenuated responses to ACh may indicate decreased NO bioavailability since ACh-mediated vasodilatation is mediated through NO, prostacyclin and endothelial-derived hyperpolarising factor(Reference Taylor, Khan, Hanson and Poston24). A reduced NO pathway is supported in the older animals where inhibition of eNOS with l-NAME had no effect on ACh-induced vasodilatation in the PR group. Moreover the enhanced responses to sodium nitroprusside are similar to those reported in eNOS knock-out mice(Reference Waldron, Ding, Lovren, Kubes and Triggle29) and in the offspring of maternal undernourished dams(Reference Holemans, Gerber, Meurrens, De Clerck, Poston and Van Assche22). Interestingly, the response to bradykinin, a second endothelial and NO-dependent vasodilator(Reference Berguer, Hottenstein, Palen, Stewart and Jacobson30), were similar between the two groups. Taken together, it is difficult to pinpoint dysfunction to any one pathway of endothelial-mediated vasodilatation.
While the exact mechanisms require further elucidation, we have demonstrated the novel finding that cardiovascular risk factors can be transmitted from the F1 to the F2 generation without additional environmental challenges. Epidemiological evidence suggests that the daughters and granddaughters of women exposed to the Dutch Hunger Winter showed decreased birth weight(Reference Painter, Roseboom and Bleker31), whilst grandparental nutrition has been shown to affect the incidence of diabetes(Reference Kaati, Bygren and Edvinsson4, Reference Pembrey, Bygren, Kaati, Edvinsson, Northstone, Sjostrom and Golding32). Animal models, too, have demonstrated transgenerational transmission of altered glucose metabolism to the F2 following a maternal dexamethasone, protein restriction or streptozotocin-induced diabetes(Reference Aerts and Van Assche3, Reference Drake, Walker and Seckl5, Reference Zambrano, Martinez-Samayoa, Bautista, Deas, Guillen, Rodriguez-Gonzalez, Guzman, Larrea and Nathanielsz6), but the present study is the first to demonstrate cardiovascular effects.
One potential route for the transgenerational transmission observed in the present study is through the F1 pregnancy. Many models of developmental programming which induced effects on the F1 offspring also alter maternal adaptations to pregnancy(Reference Ahokas, Reynolds, Anderson and Lipshitz33, Reference Brawley, Torrens, Anthony, Itoh, Wheeler, Jackson, Clough, Poston and Hanson34). We and others have demonstrated that vasodilatation is impaired in pregnant F1 offspring of protein-restricted(Reference Torrens, Brawley, Barker, Itoh, Poston and Hanson11) and total nutrient-restricted dams(Reference Hemmings, Veerareddy, Baker and Davidge35), which may have long-term consequences for the development of the F2 fetus. A second route may involve dietary effects on oocyte development. In mammals, production of the female germline stem cells is believed to cease before birth, resulting in a finite supply of germ cells(Reference Hirshfield36). In such a situation, the oocytes which gave rise to the F2 were developing in the F1 fetus during the period of protein restriction of the F0 dams. If this were so we would predict no further transmission to the F3 generation, as is seen in the altered phosphoenolpyruvate carboxykinase levels in the glucocorticoid exposed rat(Reference Drake, Walker and Seckl5).
Finally, much recent attention has focused on epigenetic changes and in particular DNA methylation and histone methylation and acetylation. Alterations in DNA methylation are heritable to future generations(Reference Morgan, Sutherland, Martin and Whitelaw37, Reference Rakyan, Chong, Champ, Cuthbert, Morgan, Luu and Whitelaw38) and can be induced in the offspring by experimental placental insufficiency and maternal protein restriction(Reference Rees, Hay, Brown, Antipatis and Palmer39–Reference Lillycrop, Phillips, Jackson, Hanson and Burdge41). Indeed, we have recently demonstrated that our model produces hypomethylation of hepatic PPARα in F1 offspring(Reference Lillycrop, Phillips, Jackson, Hanson and Burdge41) which is then transmitted to the F2 offspring(Reference Burdge, Slater-Jefferies, Torrens, Phillips, Hanson and Lillycrop8), suggesting that this could well be a method of transmission of the characteristics observed in the present study.
In conclusion, the present data demonstrate that maternal protein restriction induces raised systolic blood pressure and endothelial dysfunction in the F2 offspring, without any additional dietary challenges in the F1 offspring. Since the vascular endothelium plays an important role in the control of vascular tone, and dysfunction of the endothelium is implicated in atherosclerosis(Reference Landmesser, Hornig and Drexler13), hypertension(Reference Taddei, Virdis, Ghiadoni, Sudano and Salvetti14) and the metabolic syndrome(Reference Fornoni and Raij15), these data have implications for our understanding of heritable components of cardiovascular disease.
Acknowledgements
The present study was supported by the British Heart Foundation (M. A. H. and L. P.). Salary support for C. T. and M. A. H. was provided by the British Heart Foundation. C. T. performed the experimental work. C. T., L. P. and M. A. H. wrote the paper. There is no conflict of interest to report.




