


Diabetes type 2 is strongly associated with obesity but the mechanisms of how adipose tissue interacts with glucose metabolism are complex and still remain to be clarified. Higher BMI is associated with lower insulin sensitivity along with higher plasma levels of various adipokines, including leptin, resistin and TNF-α, which are promising candidates for linkage between insulin resistance and obesityReference Kahn and Flier1. Among the recently identified factors, the adipocyte-derived protein adiponectin is of particular interest because it may directly affect insulin-mediated glucose uptakeReference Berg, Combs, Du, Brownlee and Scherer2. Adiponectin is produced exclusively by white adipose tissue and is secreted into serumReference Scherer, Williams, Fogliano, Baldini and Lodish3. It is suggested to carry signals from adipose tissue to other organs in order to promote fat oxidationReference Hu, Liang and Spiegelman4, Reference Fruebis, Tsao, Javorschi, Ebbets-Reed, Erickson, Yen, Bihain and Lodish5and suppress hepatic glucose productionReference Berg, Combs, Du, Brownlee and Scherer2, Reference Combs, Berg, Obici, Scherer and Rosetti6. Treatment of mice with purified adiponectin significantly decreased the elevated levels of plasma NEFA, caused either by a high fat/sucrose diet or by intravenous injection of lipidsReference Fruebis, Tsao, Javorschi, Ebbets-Reed, Erickson, Yen, Bihain and Lodish5.
Several human studies revealed a strong positive correlation of fasting adiponectin and insulin sensitivity in normal subjectsReference Yamamoto, Hirose, Saito, Tomita, Taniyama, Matsubara, Okazaki, Ishii, Nishikai and Saruta7–Reference Pellme, Smith, Funahashi, Matsuzawa, Brekke, Wiklund, Taskinen and Jansson9. In obesity and type 2 diabetes, adiponectin levels are decreased, although it is produced by adipose tissueReference Hotta, Funahashi and Arita10–Reference Shea, Hilton, Orlova, Ayers and Mantzoros13. A circadian regulation of adiponectin was excluded beforeReference Shea, Hilton, Orlova, Ayers and Mantzoros13.
There are conflicting results on the effect of different meals on postprandial adiponectin levels in human subjects. Some studies found no significant difference between postprandial and fasting levelsReference Peake, Kriteros, Denyer, Campbell and Charlesworth14, Reference Imbeault, Pomerleau, Harper and Doucet15. Others found a decrease in special subgroups of subjects or after special mealsReference Esposito, Nappo, Giugliano, Di Palo, Ciotola, Barbieri, Paolisso and Giugliano16. An increase of adiponectin was even reported for obese subjectsReference English, Coughlin, Hayden, Malik and Wilding17. The associations of adiponectin gene polymorphisms with parameters of the metabolic syndrome were also inconsistent.
Because of the strong association of postprandial TAG metabolism with early insulin resistance syndromeReference English, Coughlin, Hayden, Malik and Wilding17–Reference Couillard, Bergeron, Prud'homme, Bergeron, Tremblay, Bouchard, Mauriege and Despres21 our aim was to clarify the relationship between postprandial adiponectin and TAG, insulin and glucose in a population of 110 lean and obese subjects without diabetes. We further investigated the common promoter polymorphism − 11 388 G/A for associations with fasting and postprandial parameters after a liquid fat load and a plain glucose challenge test.
Materials and methods
Subjects
Data were analysed from 110 male subjects who participated in a study aimed to evaluate postprandial metabolic values after ingestion of a fat-containing meal. Subjects were selected by age from the general population via the registration office of the town of Kiel, Germany. Inclusion criteria were age 45–65 years and absence of self-reported diabetes. Exclusion criteria were intake of hormones or lipid-lowering medication, surgery of the intestinal tract in the last 3 months or other alterations of the gastrointestinal tract, malassimilation, active cancer, chronic renal or liver disease, anaemia and alcohol abuse. All subjects gave written informed consent before participating. The study was approved by the Medical Ethical Committee of the University Clinic Kiel. Length, weight, systolic and diastolic blood pressure, heart rate and waist:hip ratio were measured. Weight was measured with an electronic scale to the nearest 0·1 kg, and height was measured to the nearest cm (Seca, Hamburg, Germany). BMI was calculated as total body weight (kg)/height (m2). For waist and hip measurements, participants were asked to roll down their undergarments. Waist circumference was measured with a constant tension tape to the nearest 0·1 cm midway between the lower rib margin and the upper iliac spine, which in most occasions was identical with the level of the umbilicus. Hip was measured at the height of trochanter femoris. Those measurements were done in an upright position with subjects breathing normally. Blood pressure was measured in a supine position after 5 min of rest by sphingomanometry (Boso, Jungingen, Germany) with the cuff at the same level as the heart during measurement. Subjects were classified as being hypertensive when treated with antihypertensive medications or their systolic/diastolic blood pressure was ≥ 130/85 mmHg. For classification of the metabolic syndrome Adult Treatment Panel IIIReference Matthews, Hosker, Rudenski, Naylor, Treacher and Turner22 criteria were used. The diagnosis of metabolic syndrome was established when three or more of the following risk factors were present: waist circumference >102 cm; TAG ≥ 150 mg/dl; HDL-cholesterol < 40 mg/dl; blood pressure ≥ 130/85 mmHg; fasting glucose >110 mg/dl.
Oral glucose tolerance test
Dietary advice was given to ensure a carbohydrate intake of >150 g/d over the previous 3 d before the test. Following a 12 h overnight fast, an intravenous catheter was inserted into a forearm vein for blood sampling and a basal blood sample was obtained. Glucose tolerance was assessed by 75 g oral glucose. Venous blood samples for glucose, insulin and adiponectin determination were taken before and 30, 60, 120, 180 and 240 min after the glucose load.
Oral metabolic tolerance test
A minimum of 3 d after the oral glucose tolerance test (oGTT) participants visited our department after a 12 hour fast. An intravenous catheter was inserted into a forearm vein for blood sampling and a fasting blood sample was obtained. The subjects drank a standardised high-fat mixed meal (500 ml) containing the following ingredients: 30 g protein (11·9 kJ%); 75 g carbohydrate (29·6 kJ%) (93 % sucrose, 7 % lactose); 58 g fat (51·6 kJ%) (65 % SFA, 35 % unsaturated fatty acids); 10 g alcohol (6·9 kJ%); 600 mg cholesterol; 30·000 IU retinylpalmitate (Nutrichem, Roth, Germany). The total energy content was 4255 kJ (1017 kcal). The test meal was drunk within 10 min after the fasting blood withdrawal. Blood withdrawal was repeated 0·5 h, 1 h, 2 h, 3 h, 4 h, 5 h, 6 h, 7 h, 8 h and 9 h after ingestion of oral metabolic tolerance test (oMTT). Subjects were allowed to walk or sit, as they wished, but not to eat or exercise during the test. Drinking water was permitted ad libitum.
Laboratory analyses
TAG, NEFA, glucose, cholesterol and HDL-cholesterol were determined using enzymatic methods with Kone lab 20i analyzer (Kone, Espoo, Finland). Plasma insulin was measured with a standard RIA (Adaltis, Freiburg, Germany). Plasma adiponectin was measured with an ELISA development kit according to the manufacturers' recommendation (R&D Systems, Minneapolis, MN, USA). All samples were measured in duplicate. The interassay and intra-assay CV were < 10 %. For the indirect determination of insulin sensitivity, the homeostasis model assessment of insulin resistance (HOMA-IR) was calculated as followsReference Matthews, Hosker, Rudenski, Naylor, Treacher and Turner22: HOMA − IR = fasting insulin (mU/l) × fasting glucose (mmol/l)/22.5. Genotyping of − 11 388 promoter polymorphism was performed by TaqMan allelic discrimination assay, using the ABI Prism 7000 sequence detection system (Applied Biosystems, Darmstadt, Germany). The probes were 5′-TTGCAAGAACCGGCTC-3′ (6-carboxyfluorescein fluorescence) and 5′-CTTGCAAGAACCAGCTC-3′ (Vaccine-type-specific probe fluorescence). The primers were 5′-GGCATCCTAAGCCCTTGCT-3′ (forward) and 5′-TTGGCAGGCTCATGTTTTGTTTT-3′ (reverse). Using the TaqMan machine, PCR conditions were denaturation at 95°C, 15 min (1 cycle) followed by 45 cycles at 95°C for 10 s and 60°C for 1 min.
Statistical analyses
All data are expressed as means and standard deviations. The 0–9 h area under the curve (AUC) was calculated by the trapezoidal method. Parameters were tested for normal distribution by the Shapiro–Wilk test. Comparisons between the groups were performed using the Mann–Whitney U test. The signed rank test was used for dependent variables. Spearman's correlation coefficient was used to determine correlation between various parameters. The Friedman test as nonparametric test for dependent variables was used for statistical evaluation of postprandial changes. We used Bonferroni-Holm correction to assess the points where significant changes occurred, thus accounting for the compounding risk of multiple statistical comparisons. The independent influence of the studied variables on adiponectin levels was tested by multiple linear regression analysis, after variables were log-transformed to achieve a normal distribution. Comparisons between the clinical characteristics of the two genotype groups were made by Mann–Whitney U test. Bonferroni-Holm correction was applied for multiple genotype-phenotype testing. A P value of < 0·05 was considered statistically significant. Δ-Adiponectin was calculated by subtracting the minimum level of postprandial adiponectin from the fasting value. The increase of NEFA was defined by the difference of NEFA maximum and NEFA nadir, the decrease of NEFA was defined by the difference of fasting NEFA and the NEFA minimum.
Results
Subjects characteristics
On average, subjects were overweight with abdominal obesity at the Adult Treatment Panel III limit23, insulin resistant referring to HOMA-normal limits ( < 2·6) defined by Monzillo & HamdyReference Monzillo and Hamdy24 and borderline hypertensive (Adult Treatment Panel III). Characteristics of the subjects are shown in Table 1. According to WHO criteria, there were nine formerly unknown type 2 diabetics among the 110 subjects. Fourteen additional subjects had either impaired fasting glucose or impaired glucose tolerance.
Table 1 General characteristics and fasting metabolic parameters of the study group (n 110)* and subjects with (n 27) or without (n 83) the metabolic syndrome (MS) according to Adult Treatment Panel III criteria
(Values are means and standard deviations)
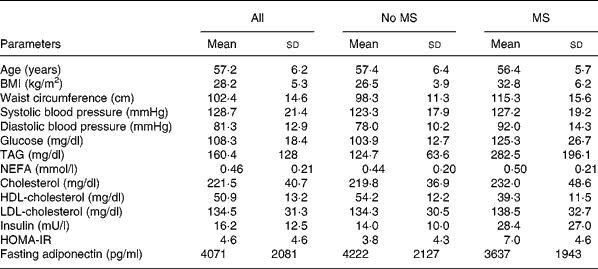
* For details of subjects and procedures, see Materials and methods.
HOMA-IR, homeostasis model assessment of insulin resistance.
Fasting and postprandial adiponectin concentrations
Adiponectin levels decreased after oGTT and after oMTT (Fig. 1). After the glucose load, levels significantly decreased from 3964 (sd 177·7) to 3744 (sd 160·7) pg/ml after 2 h by 5·9 %. After the lipid-containing meal, the decrease attained significance at 5 h and 6 h postprandial from 3964 (sd 201·3) to 3638 (sd 178·1) (P < 0·01, 8·2 %) and to 3623 (sd 175·1) (P < 0·0001, 8·6 %). After Bonferroni corrections, the decrease was still significant (Fig. 1).
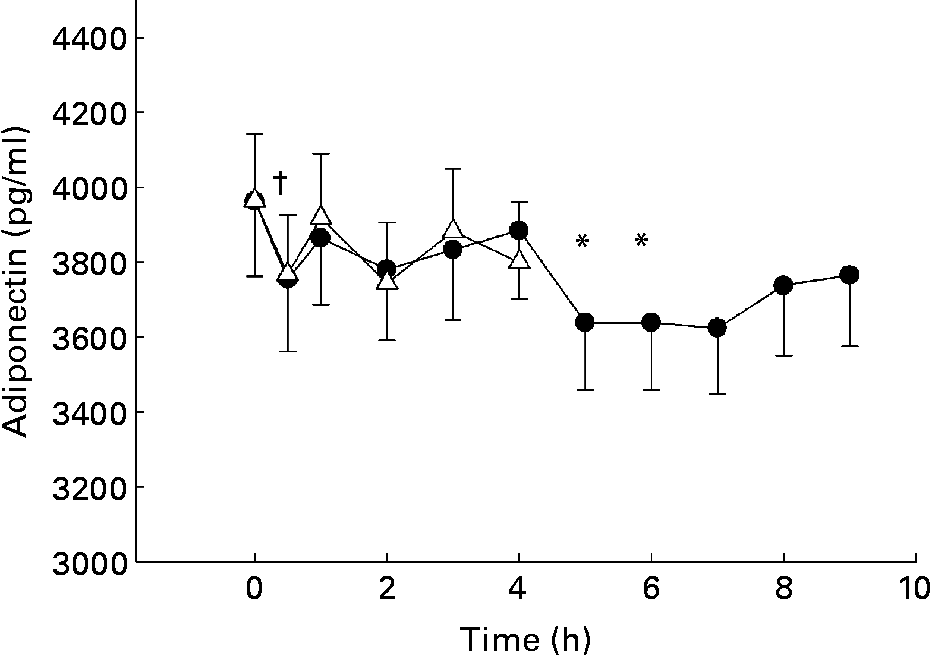
Fig. 1 Postprandial changes of adiponectin plasma levels after glucose load (oral glucose tolerance test, oGTT; Δ) and fat-containing mixed meal (oral metabolic tolerance test, oMTT; ●) in 110 subjects aged 45–65 years recruited by the town registry of Kiel and excluding known diabetes. Mean values and standard deviations. Mean values were significantly different (*P < 0·0065 after oMTT; †P < 0·017 after oGTT compared with fasting levels as assessed by Wilcoxon test after correction according to Bonferroni Holm. For details of subjects and procedures, see Materials and methods.
Fasting and postprandial (AUC) plasma adiponectin levels after oMTT and oGTT showed associations with traits of the metabolic syndrome and were negatively correlated with BMI (Tables 2 and 3). No significant correlation was found between the fasting or postprandial plasma levels of adiponectin and age found by Hotta et al. Reference Hotta, Funahashi and Arita10. The correlation with metabolic parameters after oMTT revealed a significant negative correlation of fasting and postprandial plasma adiponectin with BMI, fasting TAG, postprandial TAG and a positive correlation with HDL-cholesterol. Postprandial insulin was associated with fasting and postprandial adiponectin, whereas fasting insulin was not (Table 3). After oGTT, fasting and postprandial adiponectin levels showed a correlation with the same parameters. After OGTT, there was also a negative correlation with HOMA-IR and postprandial HOMA-IR (AUC) (Table 2).
Table 2 Correlation coefficients (r) of plasma adiponectin with clinical and metabolic parameters in the fasting state and after oral glucose tolerance test (oGTT)*
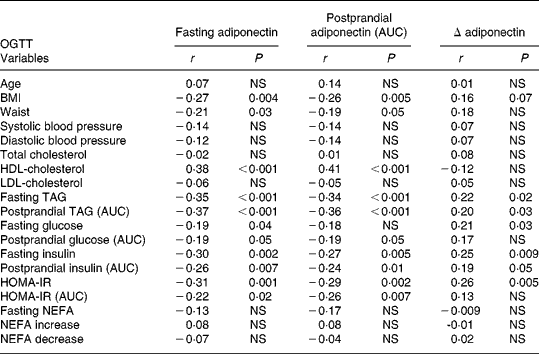
* For details of subjects and procedures, see Materials and methods.
AUC, area under the curve; HOMA-IR, homeostasis model assessment of insulin resistance; NEFA increase, NEFA maximum – NEFA nadir; NEFA decrease, fasting NEFA – NEFA minimum.
Table 3 Correlation coefficients (r) of plasma adiponectin with clinical and metabolic parameters in the fasting state and after oral metabolic tolerance test (oMTT)
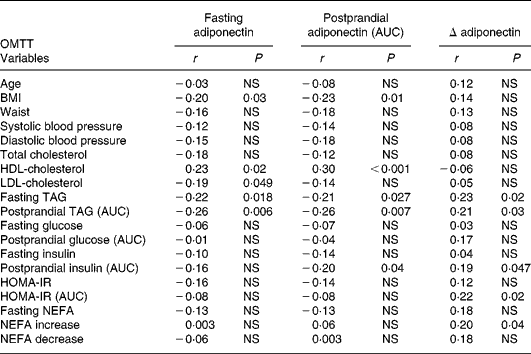
* For details of subjects and procedures, see Materials and methods.
AUC, area under the curve; HOMA-IR, homeostasis model assessment of insulin resistance; NEFA increase, NEFA maximum – NEFA nadir; NEFA decrease, fasting NEFA – NEFA minimum.
The postprandial decrease of adiponectin (Δ-adiponectin) after oGTT was correlated with the decrease of adiponectin after oMTT (r 0·24, P = 0·01). After oGTT and oMTT Δ-adiponectin was correlated with fasting adiponectin (r − 0·54, P < 0·0001; r − 0·64, P < 0·001 respectively). There was a positive correlation of Δ-adiponectin after oGTT and fasting TAG, postprandial TAG, fasting glucose, fasting and postprandial insulin (AUC) and fasting homeostasis model assessment (HOMA) (Table 2). After oMTT, Δ-adiponectin correlated with fasting and postprandial TAG, postprandial insulin, postprandial NEFA increase and postprandial HOMA (Table 3).
The findings from the afore-mentioned bivariate correlation analyses were further explored using multivariate linear analysis. We included log adiponectin as dependent variable and fasting and postprandial log glucose, log insulin, log NEFA and log TAG as independent variables. In this model, postprandial TAG (AUC) were the only independently associated variable. In another model, which included postprandial log adiponectin (AUC) as dependent variable and all the afore-mentioned factors as independent risk factors, we got the same results. Another model included log Δ-adiponectin as dependent variable and the afore-mentioned as independent. In this model, postprandial insulin, postprandial NEFA and fasting TAG remained independent predictors of Δ-adiponectin (data not shown).
Subgroup analysis
To study the effect of TAG response and insulin resistance on postprandial adiponectin course, we divided the participants into a high TAG responder group (postprandial TAG >280 mg/dl, n 47) and a normal responder group (postprandial TAG < 280 mg/dl, n 63). The insulin-resistant group was divided according to the HOMA-IR index in an insulin-resistant group (HOMA >2·6, n 70) and a group without insulin resistance (HOMA-index < 2·6, n 40)23.
The postprandial plasma adiponectin after oGTT decreased significantly in both TAG groups, but the decrease occurred later and seemed to be more prolonged in the high TAG response group (4·0 h v. 0·5 h postprandial), whereas the normal responders return to fasting level after 4 h. Insulin-resistant subjects showed no significant decrease, normal subjects had an early response at 0·5 h postprandial. The decrease of adiponectin was significantly lower in high than in normal responders and in insulin resistant than in normal subjects (Fig. 2).
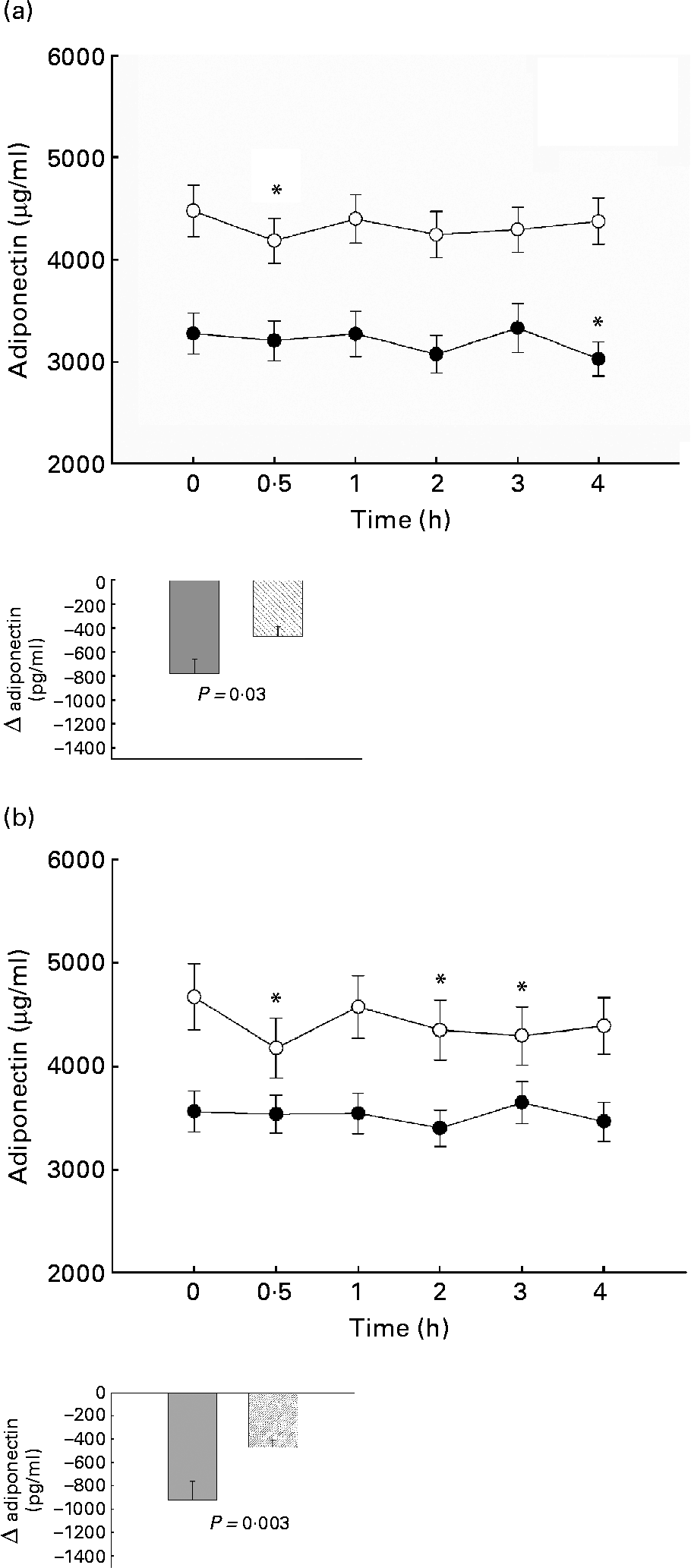
Fig. 2 Postprandial plasma adiponectin after oral glucose tolerance test in normal (○) and TAG high responders (●) (a) and in subjects with normal (○) or abnormal (●) homeostasis model assessment of insulin resistance (b). Mean values with their standard errors. *Significant decrease compared with fasting value with Bonferroni-Holm correction. For details of subjects and procedures, see Materials and methods.
After oMTT, the decrease of adiponectin did not attain statistical significance in either group. There was no significant difference in adiponectin decrease between TAG normal and high responders and subjects with or without insulin resistance.
Association of adiponectin variant −11 388 with adiponectin levels and parameters of the metabolic syndrome
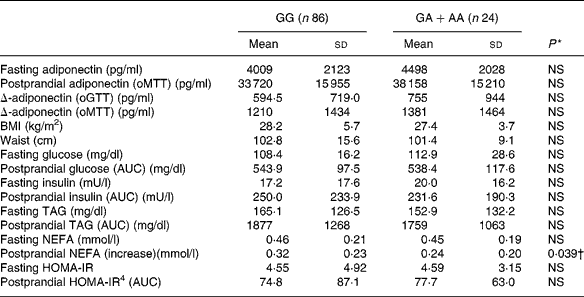
We applied a nonparametric test to all parameters for methodological consistency because most of the tested parameters did not follow a normal distribution. There was a slight, but non-significant association between adiponectin − 11 388 promoter polymorphism and adiponectin concentrations (Table 4). The single nucleotide polymorphism showed significant association with the increase of NEFA after oMTT, but after correction for multiple testing this association was not significant. There was no correlation with other parameters of the metabolic syndrome.
Table 4 Fasting and postprandial adiponectin, Δ-adiponectin (oGTT), Δ-adiponectin (oMTT) and metabolic parameters according to adiponectin −11 388 G/A promoter polymorphism‡
(Values are means and standard deviations)
* Obtained by Mann–Whitney U test.
† NS after Bonferroni Holm correction.
‡ For details of subjects and procedures, see Materials and methods.
oGTT, oral glucose tolerance test; oMTT, oral metabolic tolerance test; AUC, area under the curve; HOMA-IR, homeostasis model assessment of insulin resistance.
Discussion
The results of the present study show that plasma adiponectin decreases after a plain glucose and after a single mixed high-fat meal. These decreases were small in the complete cohort, but more pronounced in normal subjects than in subjects with traits of the metabolic syndrome and accompanying low fasting adiponectin concentrations (Fig. 2(a),(b)). These results suggest that indeed nutrient intake is followed by a decrease of adiponectin levels. However, this decrease was less pronounced in subjects with low adiponectin levels, low insulin sensitivity and high TAG. A circadian regulation of adiponectin was excluded by the authors in a study in which subjects remained awake for 38 h and received small identical snacks every 2 h. In contrast to other adipokines, such as leptin, adiponectin showed no significant circadian rhythmReference Shea, Hilton, Orlova, Ayers and Mantzoros13. In previous studies conflicting results were described for postprandial adiponectin levels with either no significant alteration or an increase of adiponectin in a small studyReference Peake, Kriteros, Denyer, Campbell and Charlesworth14, Reference Imbeault, Pomerleau, Harper and Doucet15, Reference English, Coughlin, Hayden, Malik and Wilding17, Reference Heliovaara, Strandberg, Karonen and Ebeling25. In addition to the different meal composition in those studies, the measurement of postprandial adiponectin was not performed constantly over a longer postprandial period as in the present study. Those studies might have missed the postprandial change because adiponectin showed up and down changes. Another study also used a standard liquid meal and showed a mild decrease 4 h postprandial in Prader Willi syndrome patientsReference Caixas, Gimenez-Palop, Gimenez-Perez, Potau, Berlanga, Gonzalez-Glemente, Arroyo, Laferrere and Mauricio26.
There is only one former study with a reasonable statistical power (n 60). This study investigated the response of normal and diabetic subjects to three different mealsReference Esposito, Nappo, Giugliano, Di Palo, Ciotola, Barbieri, Paolisso and Giugliano16. After the high-fat meal, diabetic and control subjects showed a significant decrease at 4 h postprandial. However, the authors did not see a significant influence of a high-carbohydrate, high-fibre meal on adiponectin. In their study, only mixed solid meals were given; thus, the effect of single nutrients remained unresolved. In the present study, we showed for the first time that not only mixed lipid-containing meals decreased adiponectin levels but also single glucose ingestion was followed by a significant decrease in adiponectin.
Although adiponectin was higher in normal than insulin-resistant subjects (Fig. 2(b)), in a multivariate analysis model, only postprandial TAG were independent predictors of fasting and postprandial adiponectin levels. This underlines the importance of postprandial TAG for fasting and postprandial adiponectin levels. The strong and independent correlation of adiponectin with TAG is supported by other studies, where the adiponectin increase after weight loss was correlated with improvement in plasma lipids independently of insulin sensitivity changesReference Baratta, Amato, Degano, Farina, Patane, Vigneri and Frittitta27.
Fasting and postprandial adiponectin after oMTT showed higher correlations with postprandial insulin and TAG than with their fasting values (Table 3). The importance of the postprandial metabolism is supported by an even stronger association of postprandial adiponectin with HDL, insulin, waist, BMI and blood pressure compared with fasting adiponectin. Interestingly, the postprandial decrease of adiponectin (Δ) itself was associated with postprandial TAG and postprandial insulin (Table 3). The common polymorphism − 11 388 in the adiponectin promoter was investigated as a genetic factor influencing adiponectin plasma levels, which was shown in some studies. These studies were epidemiological studies with large numbers of subjectsReference Fumeron, Aubert and Siddiq28, Reference Vasseur, Helbecque and Dina29. In our cohort, while we found strong associations of adiponectin plasma concentrations with several other parameters of the metabolic syndrome, we could not find significant associations of the adiponectin promoter variant with these phenotypes. The postprandial increase of NEFA after oMTT was lower in subjects with the rare allele of − 11 388 promoter polymorphism (P = 0·039) but after accounting for multiple testing this effect was not significant. We conclude that the − 11 388 promoter polymorphism has no main effect on adiponectin and parameters of the metabolic syndrome.
Our findings favour the interpretation that adiponectin has the strongest associations to postprandial lipid metabolism and is not causally related to insulin sensitivity. One could suggest that a high-fat or high-glycaemic-index diet contributes to a decrease of adiponectin levels on a long-term basis.
Acknowledgements
We thank A. Thoss, M. Hartelt, M. Gerull and S. Kaschner for excellent technical assistance. This work was financially supported by the BMBF-Project ‘Fat and metabolism – gene variation, gene regulation and gene function’ (MN 0313437A). D. Rubin and U. Helwig contributed equally to this study.






