


The metabolic syndrome is a clustering of cardiovascular risk factors, including abdominal obesity, dyslipidaemia, mitochondrial dysfunction and insulin resistance (IR)( Reference Johnson, Stenvinkel and Martin 1 ). Its prevalence is high in both developed and developing countries, where obesity and its related problems are increasing rapidly. Increased consumption of energy-dense foods containing high amounts of animal fats, SFA and fructose is thought to be the major contributor to the current epidemic of metabolic disorders( Reference Mendoza, Drewnowski and Christakis 2 ). In addition to the chronic imbalances between energy supply and energy demands, there are many studies confirming that increased availability of SFA in muscle and liver is associated with IR( Reference Hegarty, Furler and Ye 3 , Reference Boren, Taskinen and Olofsson 4 ). Muscle TAG accumulation could occur because of increased availability and uptake of systemic free fatty acids (FFA)( Reference Koh, Lee and Kim 5 ). Another possibility is that impaired muscle fatty acid oxidation (even in the presence of unaltered uptake) leads to excess cytosolic lipid accumulation( Reference Koves, Ussher and Noland 6 , Reference Rogge 7 ). The relative contribution of the different sources of excess lipid to muscle is not currently known in quantitative terms, and a combination of both factors (oversupply and impaired oxidation) may be a possibility. Although TAG accumulation is not a problem per se and can even be protective, the intracellular accumulation of long-chain CoA or various lipid derivatives is known to be disruptive. Mitochondrial FFA oxidation may influence glucose oxidation via the classic glucose–fatty acid (Randle) cycle( Reference Randle, Garland and Hales 8 ). Changes in the level of specific fatty acids could result in production of other lipid species (e.g. specific diacylglycerols (DAG)) that may alter insulin signalling pathways via protein kinase C activation( Reference Timmers, Schrauwen and de Vogel 9 , Reference Schrauwen, Schrauwen-Hinderling and Hoeks 10 ). Ceramides can also be generated from increased lipid availability and are known to be among the most active lipid second messengers to inhibit the insulin signalling pathway( Reference Hage Hassan, Bourron and Hajduch 11 ).
Mitochondria are central to energy metabolism, as they are involved in the process of oxidative phosphorylation (OXPHOS), converting the energy stored in nutrients into ATP through the action of the mitochondrial respiratory chain complexes reducing O2 to water. There is a relationship between IR and diminished mitochondrial function( Reference Montgomery and Turner 12 ). Therefore, given the central position of mitochondria and their key roles in energy metabolism and in the development of the metabolic syndrome, many therapies designed to prevent mitochondrial dysfunction have been developed( Reference Mao, Kraus and Kim 13 – Reference Smith, Hartley and Cocheme 15 ). Among these mitochondrial-targeted therapies, the antioxidant mitochondrial-targeted lipophilic ubiquinone (MitoQ) is widely used and has shown protection against oxidative damage in a wide range of pathologies( Reference Tauskela 16 , Reference Smith and Murphy 17 ), but little is known about its role in high-fat diet (HFD)-induced obesity models( Reference Gane, Weilert and Orr 18 – Reference Lim, Rashid and Jang 20 ).
This study was designed to determine the impact of MitoQ on obesity, glucose intolerance and IR development in obesogenic diet-fed rats, independent of its antioxidant action. Indeed, it is likely that MitoQ can impact on other important physiological mechanisms, particularly through direct effects on both lipid metabolism and mitochondrial activity. In line with this, it was reported that co-enzyme Q analogues may be considered as anti-adipogenic/anti-obesity factors( Reference Bour, Carmona and Galinier 21 ).
This report focused on the skeletal muscle because it is the principal tissue for postprandial insulin-stimulated glucose disposal, and thus the key regulator of glucose homoeostasis. The results suggest that MitoQ protected rats exposed to a HFD from some features of the metabolic syndrome through positive muscle lipid metabolism modulation and improved mitochondrial respiration.
Methods
Chemicals and apparatus
MitoQ was provided by R. A. J. Smith from the University of Otago in New Zealand. Peroxidase from horseradish, 2',7'-dichlorofluorescein, ADP, succinate, malate, glutamate, and oligomycin were purchased from Sigma. Rhodamine 123 was purchased from ICN Biomedical. NADPH was purchased from Calbiochem Merck. All other chemicals used were of the highest grade and obtained from Sigma Aldrich or Prolabo. LS 45 Fluorescence spectrometer (PerkinElmer) was used for fluorescence measurements. The Nicolet Evolution 100 spectrophotometer (Thermo Electron Corporation) was used for absorbance measurements.
Animals and diets
In all, twenty-four 6-week-old male Sprague–Dawley rats, weighing 175–200 g (Charles River) were housed two per cage under conditions of constant temperature (20–22°C), humidity (45–50 %) and a standard dark cycle (20.00–08.00 hours). The rats were randomised into three groups of eight animals each and were fed for 8 weeks one of the following semi-purified diets: (1) control diet with 4 % lipid as soyabean oil, (2) HFD and (3) HFD with MitoQ (HF-MitoQ). The control diet contained 20 % protein, 66 % carbohydrate, 4 % fat (soyabean oil) w/w plus cellulose and vitamin and mineral mix. The HFD contained 20 % protein, 35 % carbohydrate, 35 % fat (4 % soyabean oil+31 % lard) w/w plus cellulose and vitamin and mineral mix. MitoQ was used at a dose of 860 mg/kg diet in the form of methanesulfonate β-cyclodextrin complex, which corresponded to 375 µmol of MitoQ/kg diet. Rats were given free access to tap water and food. Body weight and food consumption were determined weekly. Our institution guidelines for the care and use of laboratory animals were observed, and all the experimental procedures were approved by the local ethics committee (reference CEEA-LR-12002). This report is part of a larger study where the effects of other antioxidants (apocynin, allopurinol and N-acetyl-cysteine) have been investigated and published elsewhere( Reference Feillet-Coudray, Fouret and Ebabe Elle 22 ).
Oral glucose tolerance test
The oral glucose tolerance test (OGTT) was performed as previously described( Reference Aoun, Fouret and Michel 23 ). In brief, on week 7 of diet intake, after 16 h of fasting, rats received by gavage 2 g glucose/kg body weight. Blood was sampled through the tail vein of conscious rats immediately before the gavage and 20, 40, 60, 90, 120 and 180 min afterward, and the blood glucose level was measured using glucose strips and a commercial glucometer (Accu-Chek Active; Roche Diagnostics). The AUC values were calculated by the trapezium method. The AUC values are expressed as mmol glucose/l per s.
Killing of rats and metabolic biochemical analyses
After 4–5 d of the OGTT, after 16 h of fasting, rats were anaesthetised with pentobarbital (40 mg/kg body weight) (Ceva Santé Animale), and blood samples were collected from the abdominal artery with a heparinised syringe and distributed into heparinised and dry tubes. Blood tubes were centrifuged at 1000 g for 10 min at 4°C, and plasma or serum samples were collected and stored at −80°C until analysis. Both gastrocnemius were removed and one of them was used for the extemporaneously isolation of fresh muscle mitochondria. The other gastrocnemius as well as the soleus muscles and the liver and the visceral fat were collected, rinsed and weighed.
Plasma glucose, serum total cholesterol, TAG and NEFA levels were measured by enzymatic techniques (Thermo Electron Corp.). Plasma insulin, leptin, TNF-α and IL-6 levels were quantified using ELISA kits (St Charles). IR was evaluated by the homoeostasis model assessment of IR (HOMA-IR) formula: (fasting plasma glucose level (mmol/l)×fasting plasma insulin level (mIU/l))/22·5( Reference Pu, Gao and Mohamed 24 ).
Muscle expression of TNF-α and IL-6 cytokines
The muscle mRNA expressions of TNF-α and IL-6 genes were determined by RT-quantitative PCR as previously reported( Reference Feillet-Coudray, Fouret and Ebabe Elle 22 ). Normalisations were made to RPS9. The following primer sequences were used: forward TNF-α – GGC ATG GAT CTC AAA GAC AAC C; reverse TNF-α – CAA ATC GGC TGA CGG TGT G (accession no. NM_012675.3); forward IL-6 – CTC CGG ACT TGT GAA GTA GGG; reverse IL-6 – ATG AAG TTT CTC TCC GCA AGA (accession no. NM_012589.1); forward RPS9 – GAA GCT GGG TTT GTC GCA AA; and reverse RPS9 – CGG AGC CCA TAC TCT CCA AT (accession no. NM_031108.4). Western blotting analyses for determining muscle TNF-α and IL-6 protein expressions were carried out as previously reported( Reference Feillet-Coudray, Fouret and Ebabe Elle 22 ). β-Actin was used as reference, and blot intensities were measured using ImageJ software (NIH). Antibodies of TNF-α (ab66579) and IL-6 (ab6672) were from Abcam and β-actin antibody (s-c81178) was from Santa Cruz Biotechnology.
Mitochondrial membrane potential, respiration and reactive oxygen species production
Muscle mitochondria were isolated by the differential centrifugation technique( Reference Aoun, Feillet-Coudray and Fouret 25 ). The protein content of muscle supernatants and mitochondrial suspensions was determined according to Bradford using bovine serum albumin as the standard( Reference Bradford 26 ).
Mitochondrial membrane potential was monitored according to the method described by Baracca et al.( Reference Baracca, Sgarbi and Solaini 27 ) using rhodamine as a probe. Mitochondrial potential (Δψ) change has been evaluated by measuring rhodamine fluorescence at the following steps: with rhodamine label alone (2 µm) in MIRO5 buffer plus muscle fresh mitochondria (0·25 mg proteins), plus glutamate–malate–succinate (2·5:5:5 m), plus ADP (0·5 mm). RH-123 fluorescence was measured at 495 nm (excitation) and 525 nm (emission) under continuous stirring.
Mitochondrial respiration was determined by measuring mitochondrial VO2 in two 2-ml air-tight thermo-stated chambers of high resolution Oxygraph (Oxygraph2k; Oroboros)( Reference Aoun, Feillet-Coudray and Fouret 25 ). The chambers were equilibrated with the MIRO5 respiration buffer at 37°C and fresh mitochondria were then added (0·4 mg protein) followed by substrates malate–glutamate–succinate (2·5:5:5 mm, state 4) and ADP 0·5 mm (state 3). Respiratory coefficient ratio was calculated as the state 3:state 4 ratio.
Mitochondrial reactive oxygen species (ROS) production was measured on fresh mitochondria as previously described with minor modifications( Reference Capel, Rimbert and Lioger 28 ). In brief, reaction mixtures (2 ml), containing MIRO5 buffer, dihydrodichlorofluorescein (10 µm) and peroxidase (2 IU/ml), were incubated with 60 µg of mitochondrial proteins with or without substrates (malate–glutamate–succinate, 2·5:5:5 mm, respectively) and with or without ADP (0·5 mm). Fluorescence was measured 30 min after reaction initiation at 490/525 nm excitation and emission, respectively.
Measurement of mitochondrial enzyme activities
Citrate synthase (CS) was used as the mitochondrial marker and measured in the muscle supernatants according to Srere( Reference Srere 29 ). The activities of the four mitochondrial respiratory complexes (complex I, complex II, complex II+III and complex IV) were determined spectrophotometrically( Reference Aoun, Fouret and Michel 23 ). ATPase activity (complex V) was determined as described by Teodoro et al.( Reference Teodoro, Rolo and Duarte 30 ) with minor modifications( Reference Aoun, Fouret and Michel 23 ). Moreover, enzymatic activity of mitochondrial medium-chain acyl-CoA dehydrogenase (MCAD) and β-hydroxyacyl-CoA dehydrogenase (β-HAD) was determined spectrophotometrically( Reference Djouadi, Riveau and Merlet-Benichou 31 , Reference Clayton, Eaton and Aynsley-Green 32 ). Mitochondrial aconitase, reported as a good marker of oxidative stress in oxidative pathologies, was determined on muscle supernatants( Reference Nulton-Persson and Szweda 33 ).
Mitochondrial oxidative phosphorylation subunits
The OXPHOS Western blotting kit (MitoSciences) containing a cocktail of monoclonal antibodies was used to semi-quantify the relative levels of the subunits of the mitochondrial OXPHOS complexes: NDUFB8 20 kDa of complex I, Fe–S 30 kDa of complex II, core 2–47 kDa of complex III and α 53 kDa subunit of ATP synthase( Reference Flamment, Rieusset and Vidal 34 ). For detection of OXPHOS subunits, all Western blot steps were followed.
Muscle lipid analysis
Muscle samples were homogenised in NaCl (9 g/l) and Triton X-100 (0·1 %), and levels of FFA, TAG and total cholesterol were quantified on the muscle homogenate by enzymatic methods (Wako NEFA-C kit; Oxoid; Cholesterol RTU kit and triglycerides PAP kit; Biomérieux).
Muscle ceramide content and enzymes of ceramide metabolism
The muscle content of ceramide species (C14 : 0, C16 : 0, C18 : 0, C18 : 1, C20 : 0, C24 : 0, C24 : 1) was determined by means of ultra-HPLC according to Blachnio-Zabielska et al.( Reference Blachnio-Zabielska, Persson and Koutsari 35 ). The activities of the key enzymes implicated in ceramide metabolism – serine palmitoyltransferase (SPT) and neutral sphingomyelinases (nSMase) and acidic sphingomyelinases), enzymes responsible for ceramide generation, and neutral ceramidase (nCDase) and acidic ceramidase, enzymes responsible for ceramides degradation – were measured using radiolabelled substrates with liquid scintillation counter as previously described( Reference Blachnio-Zabielska, Zabielski and Baranowski 36 ).
Muscle diacylglycerol content in muscle lipid extracts by TLC-densitometry
Muscle lipids were extracted using a mixture of chloroform–methanol (2:1, v/v) in the presence of butylated hydroxytoluene (50 mg/l). The application of lipid extracts of muscle homogenate on silica gel 60 HPTLC plates (250 µm, 20×10 cm; Merck), pre-treated with 1·5 % p/v boric acid in ethanol (100 %), was automatically performed on a 4-mm band width using a CAMAG ATS4 apparatus. Two developments were performed, first with pentane–chloroform–methanol (52:45:3, by volume) on 60-mm migration distance and second with pentane–diethyl ether (97:3 by volume) on 67-mm migration distance, which allowed the separation of neutral lipids. The scanning of the plates was carried out using a CAMAG TLC scanner 3 operating in the reflectance mode. The plates were scanned at 550 nm after dipping in a solution of copper sulphate 640 mm in H3PO4 1·18 m and heating for 20 min at 180°C. The 1-monoacylglycerol (MAG), 1,2-DAG and 1,3-DAG contents were finally identified by comparing their retention factor (R f ) with authentic standards and were quantified using calibration curves of the same standards.
Phospholipid classes in muscle lipid extracts by TLC-densitometry
Muscle lipids were extracted using a mixture of chloroform–methanol (2:1, v/v) in the presence of butylated hydroxytoluene (50 mg/l). P was quantified to determine total muscle phospholipids( Reference Bartlett 37 ). The application of muscle chloroform–methanol extracts on silica gel 60 HPTLC plates (250 µm, 20×10 cm), pre-treated with 2·3 % p/v boric acid in ethanol (100 %), was automatically performed on a 4-mm band width using a CAMAG ATS4 apparatus. The development was performed with methanol–acetic acid–pentane–chloroform (15:10:30:45, by volume), which allowed the separation of phospholipids and neutral lipids on a 60-mm total migration distance. The scanning of the plates was carried out using a CAMAG TLC scanner 3, operating in the reflectance mode. The plates were scanned at 715 nm after dipping in a solution of Blue Spray (1:2:3, v/v/v, Blue Spray–H2SO4 4·2 m–acetone) and heating for 3 min at 55°C. The different classes of phospholipids (phosphatidylcholine (PC); phosphatidylethanolamine (PE); phosphatidylinositol (PI); phosphatidylserine (PS); sphingomyelin (SM); lysophosphatidylcholine (LPC); phosphatidylglycerol (PG); phosphatidic acid (PA) and cardiolipin (CL)) were finally identified by comparing their R f with authentic standards and were quantified using calibration curves of the same standards( Reference Macala, Yu and Ando 38 ).
Fatty acid composition of muscle and muscle phospholipids
Determination of fatty acid composition of the total muscle lipids was carried out using the muscle chloroform–methanol extracts. To determine the fatty acid composition of the muscle phospholipids, the phospholipid fraction was separated from other muscle neutral lipids on TLC plates with hexane–diethyl ether–glacial acetic acid mixture (70:30:1, v/v/v). Plates were dried and sprayed with dichlorofluorescein. Bands corresponding to phospholipids were identified under UV and collected by scraping the silica into a glass tube for further analysis. The dried muscle chloroform–methanol extract and the scraped phospholipid bands were trans-esterified with an alkali mixture of KOH–methanol for 10 min at room temperature. The total fatty acid methyl ester pattern was analysed by GC (Agilent Technologies) equipped with a DB-23 column and a flame ionisation detector. Chromatograms were collected, and peaks were integrated and identified by comparison with commercially available references.
Statistical analysis
The sample size was calculated from the expected difference in the AUC of the OGTT between the control group and the HFD group. We expected an AUC about 1·20 times greater in the HF group than in the control group. A standard deviation of 15 % of the higher value could be allowed. For a type 1 risk α of 0·05 and power (1-β) of 80 %, the sample size required was eight per group.
Data are expressed as mean values and standard deviations. Normal distribution of data was checked by the Kolmogorov–Smirnov test. To detect statistical significance among groups, one-way ANOVA test, followed by Fischer’s post hoc analysis, was applied for normally distributed data. Data not following normal distribution were analysed by non-parametric tests using the Kruskal–Wallis method followed by Dunn’s post hoc analysis and marked with †. The limit of statistical significance was set at P<0·05. Statistical analyses were performed using the StatView program (SAS Institute).
Results
Mitochondrial-targeted lipophilic ubiquinone intake prevented weight gain, glucose intolerance and muscle lipid accumulation
Although the dietary intake (g/d) was decreased, the energy intake was significantly increased in the HF group but not in the MitoQ group (Table 1). Therefore, the final rat body weight was significantly increased in the HF group compared with the control group, whereas MitoQ intake prevented weight gain (Fig. 1). Moreover, the weight of both liver and adipose tissue was increased in the HF groups, but this increase was largely attenuated by MitoQ intake. However, the weight of both gastrocnemius and soleus muscles remained unchanged among the studied groups (Table 1).

Fig. 1 Body weight gain in rats fed either (1) control (cont, ![]() ), (2) high fat (HF,
), (2) high fat (HF, ![]() ) or (3) HF+MitoQ (
) or (3) HF+MitoQ (![]() ) diets for 8 weeks. Values are means (n 7–8 observations), and standard deviations. Statistical significance among groups was detected as described in the statistical analysis section. The limit of statistical significance was set at P<0·05. In all, twenty-four 6-week-old male Sprague–Dawley rats, weighing 175–200 g, were randomised into three groups of eight animals each and fed their corresponding diets for 8 weeks. Rats were given free access to tap water and food, and body weight and food consumption were determined weekly. MitoQ, mitochondrial-targeted lipophilic ubiquinone; HF+MitoQ, HF with MitoQ.
) diets for 8 weeks. Values are means (n 7–8 observations), and standard deviations. Statistical significance among groups was detected as described in the statistical analysis section. The limit of statistical significance was set at P<0·05. In all, twenty-four 6-week-old male Sprague–Dawley rats, weighing 175–200 g, were randomised into three groups of eight animals each and fed their corresponding diets for 8 weeks. Rats were given free access to tap water and food, and body weight and food consumption were determined weekly. MitoQ, mitochondrial-targeted lipophilic ubiquinone; HF+MitoQ, HF with MitoQ.
Table 1 Effects of diets on diet consumption, rat body evolution and tissue weightsFootnote * (Mean values and standard deviations; n 6–8 observations)
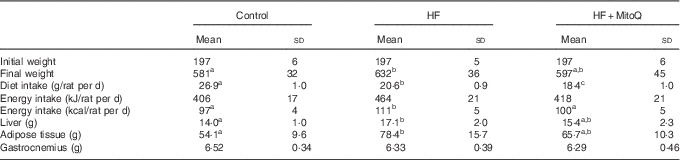
HF, high fat.
a,b,cMean values with unlike superscript letters in the same row were significantly different. The limit of statistical significance was set at P<0·05.
* Rats were fed their corresponding diets for 8 weeks; 16-h fasted rats were killed and liver, muscles and adipose tissues were sampled, rinsed with physiological solution, weighed and aliquoted and frozen for analysis. To detect statistical significance among groups, one-way ANOVA test, followed by Fischer’s post hoc analysis, was applied for normally distributed data checked by the Kolmogorov–Smirnov test.
Two major inflammation markers, TNF-α and IL-6, were determined in this study. Plasma TNF-α was increased significantly in the HF group and increased non-significantly in the HF-MitoQ group compared with the control group. However, plasma IL-6 remained unchanged in all the three experimental groups. In addition, the gene and protein expressions of both TNF-α and IL-6 remained unchanged at the muscle level (Table 2). Although the fasted plasma glucose level remained unchanged among the studied groups, the AUC of the OGTT was increased in the HF group, whereas MitoQ intake attenuated it compared with the control group (Fig. 2). In addition, both plasma insulin level and the HOMA-IR index were decreased by MitoQ intake v. both control and HF groups, whereas plasma leptin was increased in both HF and HF-MitoQ groups v. the control group (Table 2). Surprisingly, some serum lipid parameters (TAG, NEFA and total cholesterol) were decreased significantly in the HF rat group and MitoQ intake further lowered these parameters. The muscle TAG level was statistically significantly increased (+23 %) in the HF group v. the control group, whereas MitoQ intake completely prevented this muscle lipid accumulation (Table 2).

Fig. 2 Glucose tolerance test in 16-h fasted rats fed either control (cont, ![]() ), high-fat (HF,
), high-fat (HF, ![]() ) or HF+MitoQ (
) or HF+MitoQ (![]() ) diets for 8 weeks. Values are means (n 7–8 observations), and standard deviations represented by vertical bars. Statistical significance among groups was detected as described in the statistical analysis section. The limit of statistical significance was set at P<0·05. a,bGroup mean values with unlike letters were significantly different. In all, twenty-four 6-week-old male Sprague–Dawley rats, weighing 175–200 g, were randomised into three groups of eight animals each and fed their corresponding diets for 8 weeks. Rats were given free access to tap water and food, and body weight and food consumption were determined weekly. MitoQ, mitochondrial-targeted lipophilic ubiquinone; HF+MitoQ, HF with MitoQ.
) diets for 8 weeks. Values are means (n 7–8 observations), and standard deviations represented by vertical bars. Statistical significance among groups was detected as described in the statistical analysis section. The limit of statistical significance was set at P<0·05. a,bGroup mean values with unlike letters were significantly different. In all, twenty-four 6-week-old male Sprague–Dawley rats, weighing 175–200 g, were randomised into three groups of eight animals each and fed their corresponding diets for 8 weeks. Rats were given free access to tap water and food, and body weight and food consumption were determined weekly. MitoQ, mitochondrial-targeted lipophilic ubiquinone; HF+MitoQ, HF with MitoQ.
Table 2 Effects of diets on blood and muscle lipid, inflammation and insulin-resistance parametersFootnote * (Mean values and standard deviations; n 6–8 observations)

HF, high fat; HOMA-IR, homoeostasis model assessment-insulin resistance; ww, wet weight; FFA, free fatty acids.
a,b Mean values with unlike letters in the same row were significantly different. The limit of statistical significance was set at P<0·05.
* Rats were fed their corresponding diets for 8 weeks; 16-h fasted rats were killed and blood and muscles were sampled. The muscle was rinsed with physiological solution, weighed and aliquoted and frozen for analysis. To detect statistical significance among groups, one-way ANOVA test, followed by Fischer’s post hoc analysis, was applied for normally distributed data checked by the Kolmogorov–Smirnov test.
† Data not following normal distribution were analysed by non-parametric tests using the Kruskal–Wallis method followed by Dunn’s post hoc analysis.
Mitochondrial-targeted lipophilic ubiquinone intake restored mitochondrial respiration
The mitochondrial membrane potential and the mitochondrial ROS production measured ex vivo were not affected by the HFD or MitoQ intake (Table 3). However, mitochondrial respiration decreased significantly with the HF diet v. control diet in the three studied conditions: mitochondria challenged with glutamate–malate or with glutamate–malate–succinate or with G/M/S plus ADP. MitoQ intake significantly increased mitochondrial respiration back to control values (Fig. 3). However, mitochondrial CS activity, enzymatic activity and the protein expression of mitochondrial chain complexes also remained unchanged in the studied groups (Table 3). Finally, mitochondrial β-oxidation was investigated by assessing the enzymatic activity of two major enzymes (β-HAD and MCAD) in this pathway. The activity of these two enzymes remained unchanged in the studied groups (Table 3).

Fig. 3 Muscle mitochondrial respiration in rats fed either control (cont), high-fat (HF) or HF+MitoQ diets for 8 weeks. Values are means (n 7–8 observations), and standard deviations represented by vertical bars. Rats were fed their corresponding diets for 8 weeks; 16-h fasted rats were killed and muscles were sampled, rinsed with physiological solution and fresh mitochondria were isolated. (A) Mitochondria were challenged by malate + glutamate. (B) Mitochondria were challenged by malate+glutamate+succinate. (C) Mitochondria were challenged by malate+glutamate+succinate plus ADP. (D) Respiratory coefficient ratio (state 3/state 4). To detect statistical significance among groups, a one-way ANOVA test, followed by Fischer’s post hoc analysis, was applied for normally distributed data checked by the Kolmogorov–Smirnov test. The limit of statistical significance was set at P< 0·05. a,bMean values with unlike letters for the same parameter were significantly different. † Data not following normal distribution were analysed by non-parametric tests using the Kruskal–Wallis method followed by Dunn’s post hoc analysis. MitoQ, mitochondrial-targeted lipophilic ubiquinone; HF+MitoQ, HF with MitoQ.
Table 3 Effects of diets on mitochondrial activityFootnote * (Mean values and standard deviations; n 6–8 observations)

HF, high fat; ROS, reactive oxygen species; G, glutamate; M, malate; S, succinate; MCAD, medium-chain acyl-CoA dehydrogenase; β-HAD, β-hydroxyacyl-CoA dehydrogenase.
The limit of statistical significance was set at P<0·05. There were no significant differences among the three experimental groups for all the stated parameters.
* Rats were fed their corresponding diets for 8 weeks; 16-h fasted rats were killed and muscles were sampled, rinsed with physiological solution, weighed and aliquoted and frozen for analysis. To detect statistical significance among groups, one-way ANOVA test, followed by Fischer’s post hoc analysis, was applied for normally distributed data checked by the Kolmogorov–Smirnov test.
Mitochondrial-targeted lipophilic ubiquinone intake was without effect on diacylglycerol content, but decreased muscle enzymatic activities of ceramide metabolism
The muscle MAG as well as DAG contents were not significantly affected by either the HF diet or MitoQ intakes (Table 4). However, the HF diet intake trended to increase the muscle content of both 1,2-DAG (+15 %) and 1,3-DAG (+22 %), whereas MitoQ tended to decrease both DAG contents back to the control values (−10 and −31 %, respectively).
Table 4 Muscle diacylglycerol (DAG) and ceramide contents and enzymes of ceramide metabolismFootnote * (Mean values and standard deviations; n 6–8 observations)

HF, high fat; MAG, 1-monoacylglycerol.
a,b Mean values with unlike letters in the same row were significantly different. The limit of statistical significance was set at P<0·05.
* Rats were fed their corresponding diets for 8 weeks; 16-h fasted rats were killed and muscles were sampled, rinsed with physiological solution, weighed and aliquoted and frozen for analysis. To detect statistical significance among groups, one-way ANOVA test, followed by Fischer’s post hoc analysis, was applied for normally distributed data checked by the Kolmogorov–Smirnov test.
† Data not following normal distribution were analysed by non-parametric tests using the Kruskal–Wallis method followed by Dunn’s post hoc analysis.
The total content of muscle ceramides did not change by the HF diet or MitoQ intake. However, some species of ceramides (C14, C16, C18 : 1, C22 and C24 : 1) were significantly decreased by HF diet intake while MitoQ administration was without effect. The major ceramide species, the C18, as well as the C20 and C24, remained unchanged in the three studied groups (Table 4).
The activities of the key enzymes implicated in ceramide and SM metabolism showed that SPT activity was decreased non-significantly (−14 %) in both HF and HF-MitoQ diets v. control diet, whereas the activity of both nCDase and nSMase were increased only in the HF diet v. control diet. Interestingly, the MitoQ intake decreased significantly the activity of these two enzymes back to control values (Table 4).
Mitochondrial-targeted lipophilic ubiquinone intake decreased the percentage of sphingomyelin in the muscle
The major finding from the muscle phospholipid analysis was the significant increase in the SM percentage and in the SM:PC ratio with the HF diet compared with the control diet. Interestingly, MitoQ intake was proved to be efficient in normalising SM per cent and SM:PC ratio to control levels (Table 5). The percentage of the other phospholipid classes (LPC, PC, PI, PS, PE, PG, PA and CL) was not altered by either the HF diet or MitoQ intake.
Table 5 Effects of diets on muscle phospholipid content and their class distributionFootnote * (Mean values and standard deviations; n 6–8 observations)
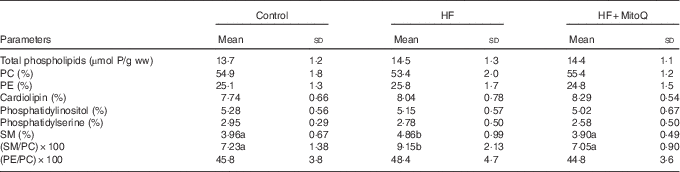
HF, high fat; ww, wet weight; PC, phosphatidylcholine; PE, phosphatidylethanolamine; SM, sphingomyelin.
a,b Mean values with unlike letters in the same row were significantly different. The limit of statistical significance was set at P<0·05.
* Rats were fed their corresponding diets for 8 weeks; 16-h fasted rats were killed and muscles were sampled, rinsed with physiological solution, weighed and aliquoted and frozen for analysis. To detect statistical significance among groups, one-way ANOVA test, followed by Fischer’s post hoc analysis, was applied for normally distributed data checked by the Kolmogorov–Smirnov test.
Mitochondrial-targeted lipophilic ubiquinone intake increased muscle PUFA n-3 %
We have analysed the effects of the HF diet and MitoQ intake on the fatty acid composition of total muscle lipids (Table 6). The percentage of total and individual SFA (C16 : 0 and C18 : 0) remained unchanged. Apart from this, the HF diet intake was associated with a decrease in 16 : 1 n-7 and 18 : 1 n-7 and with an increase in 18 : 1 n-9 % (+100 %), and as a consequence a strong tendency for an increase in the level of total MUFA (+45 %). The MitoQ intake only slightly decreased the 18 : 1 n-9 % (−20 %) and the total MUFA percentage (−18 %) in the total muscle lipid compared with the HF diet. The HF diet intake also increased non-significantly the C22 : 5 n-3 % and decreased significantly the C22 : 6 n-3 %, with an overall decrease in the percentage of total PUFA n-3 in the total muscle lipid (Table 6). Interestingly, MitoQ intake increased significantly the total PUFA n-3 % (+28 %), particularly that of C22 : 5 n-3 (+19 %) and C22 : 6 n-3 (+25 %), compared with the HF diet (Table 6).
Table 6 Effects of diets on fatty acid composition (%) of total lipids from muscle homogenateFootnote * (Mean values and standard deviations; n 6–8 observations)

HF, high fat.
a,b Mean values with unlike letters in the same row were significantly different. The limit of statistical significance was set at P<0·05.
* Rats were fed their corresponding diets for 8 weeks; 16-h fasted rats were killed and muscles were sampled, rinsed with physiological solution, weighed and aliquoted and frozen for analysis. To detect statistical significance among groups, one-way ANOVA test, followed by Fischer’s post hoc analysis, was applied for normally distributed data checked by the Kolmogorov–Smirnov test.
† Data not following normal distribution were analysed by non-parametric tests using the Kruskal–Wallis method followed by Dunn’s post hoc analysis.
Mitochondrial-targeted lipophilic ubiquinone intake decreased the percentage of MUFA in the muscle phospholipids
The HF diet and MitoQ intake were without effect on the percentage of total or individual SFA in muscle phospholipids, although the per cent of C18 : 0 was non-significantly increased (+15 %) with HF diet and MitoQ intake (Table 7). On the contrary, the MUFA per cent was highly impacted. The HF diet and MitoQ intake were associated with a significant decrease in the percentages of 16 : 1 n-7 and 18 : 1 n-7, with a strong tendency for a decrease in total MUFA per cent with MitoQ intake (−20 %) (Table 7). The HF diet and MitoQ intake did not affect the PUFA n-6 %, although the per cent of C20 : 4, n-6 was increased non-significantly and significantly with HF diet and MitoQ, respectively. The HF diet intake decreased significantly the PUFA n-3 %, whereas MitoQ intake slightly prevented this decrease. Indeed, HF diet and MitoQ intake increased the C22 : 5 n-3 % and decreased the C22 : 6 n-3 % (Table 7).
Table 7 Effects of diets on fatty acid composition (%) of muscle phospholipidsFootnote * (Mean values and standard deviations; n 6–8 observations)

HF, high fat.
a,b Mean values with unlike letters in the same row were significantly different. The limit of statistical significance was set at P<0·05.
* Rats were fed their corresponding diets for 8 weeks; 16-h fasted rats were killed and muscles were sampled, rinsed with physiological solution, weighed and aliquoted and frozen for analysis. To detect statistical significance among groups, one-way ANOVA test, followed by Fischer’s post hoc analysis, was applied for normally distributed data checked by the Kolmogorov–Smirnov test.
† Data not following normal distribution were analysed by non-parametric tests using the Kruskal–Wallis method followed by Dunn’s post hoc analysis.
Discussion
We have investigated the impact of MitoQ, on the development of the metabolic syndrome features in obesogenic diet-fed rats. The results of this study show that the HF diet led to a moderate gain in weight and glucose intolerance associated with slight low-grade inflammation, intramuscular lipid accumulation, membrane phospholipid modification and alteration in muscular mitochondrial respiration. In particular, the HF diet significantly increased the muscle levels of both TAG and SM and significantly decreased the muscle PUFA n-3 level. In addition, these HF diet-induced lipid modifications were accompanied with significant decrease in muscle mitochondrial respiration. Interestingly, MitoQ intake prevented body weight gain, improved glucose tolerance, corrected the observed lipid alterations and restored mitochondrial respiration. These results suggest that muscle lipid modulation and mitochondrial respiration restoration by MitoQ intake may mediate the prophylactic action of MitoQ in some metabolic syndrome features in HFD-fed rats. Although our results show that MitoQ intake was accompanied by slight but significant decrease in food and energy intakes (about −10 %) compared with the HF group, the food consumption was rather stable during the whole experimental period, and thus the cause for this food intake decrease did not seem to be due to food aversion. The mechanism involving decreased energy intake with MitoQ may be mediated at the hypothalamic level as reported by Fink et al.( Reference Fink, Herlein and Guo 39 ) for the effects of quinols in mice fed HFD.
Low-grade inflammation is a characteristic of the obese state, and adipose tissue is known to release many inflammatory mediators. In this study, the HF intake was accompanied with significant increase in plasma TNF-α, but not in IL-6 levels, whereas MitoQ only slightly attenuated this increase. However, muscle gene and protein expressions of these two cytokines were not altered by either the HF diet or MitoQ intake. It is highly likely that the source of this increased plasma TNF-α level is the adipose tissue, which is known to be the major sources of body cytokines.
One intriguing finding is that MitoQ intake decreased the plasma insulin level compared with both control and HF diet groups. This may suggest that MitoQ somehow increased insulin sensitivity. The decreased insulin levels could also suggest a decreased glucose-stimulated insulin release at the pancreas level. However, as we did not sample the pancreas of these animals in the present study, these hypotheses will be addressed in future studies.
Because of the key role of mitochondria in lipid metabolism, obesity and IR, and because of the characteristics of MitoQ as a mitochondrial-targeted quinone, we have determined many surrogate parameters to evaluate muscle mitochondrial function and activity in this study. Both HF diet and MitoQ intake showed no notable effect on mitochondrial biogenesis as determined by CS activity and mitochondrial DNA. Ex vivo mitochondrial membrane potential, ROS production and activity and protein content of mitochondrial respiratory complexes, as well as mitochondrial β-oxidation activity, as measured by β-HAD and MCAD activities also remained unchanged in the three studied groups. In addition, there were no significant changes in either protein expressions or enzymatic activities of the mitochondrial respiratory chain complexes. However, the muscle mitochondrial respiration/VO2 was decreased with the HF diet and was reestablished to the control value by MitoQ intake. The MitoQ intake restored this mitochondrial respiration probably by both modulating the fatty acid composition of mitochondrial membranes and increasing the mitochondrial quinone pool. Indeed, the later is involved as an electron and proton carrier in mitochondrial respiration, and can act in its reduced form as an antioxidant improving mitochondrial respiration as reported by Jiménez-Santos et al.( Reference Jiménez-Santos, Juarez-Rojop and Tovilla-Zarate 40 ). This enhanced mitochondrial respiration may lead to higher fat oxidation, and thus to changes in intramuscular lipid content and composition, which may mediate the prophylactic action of MitoQ on some aspects of the metabolic syndrome. However, one limitation of ex vivo measurement of mitochondrial respiration is that the muscle mitochondria were not in their natural environment, and thus we should be cautious in the interpretation of these particular results.
Muscle lipid accumulation and/or lipid metabolism alteration is thought to play an important role in the development of IR in HF feeding models( Reference van Herpen and Schrauwen-Hinderling 41 , Reference Muoio 42 ). Although many reports have reported increased muscle lipid accumulation in mice/rats fed HFD, the intracellular toxic metabolites (fatty acyl CoA, DAG, ceramides and SM) are considered to be the true culprits of IR that may impair insulin signalling to glucose metabolism( Reference Muoio 42 – Reference DeFronzo 44 ). In line with this, the results of this study show that the muscle lipid profile is altered in our obesogenic diet-fed rats. The HF intake has altered different major actors of lipid metabolism and signalling – that is, (1) an increase in activities of key enzymes implicated in muscle ceramide metabolism; (2) an increase in muscle SM % and (3) a decrease in the PUFA n-3 % with the HF diet. Interestingly, the MitoQ intake has led to the normalisation of these parameters nearly to the control values.
The HF diet intake was accompanied by a slight decrease in total ceramide content (−9·5 %) and led to a significant decrease in selective ceramide species. This may arise from diminished de novo ceramide synthesis (slight low SPT activity in both HF diet groups), and from its increased degradation by activation of nCDase. However, the high sphingomyelinase (SMase) activity can also balance the high nCDase activity; thus, ceramide does not highly accumulate in skeletal muscle due to its faster turnover rate. Although MitoQ intake decreased significantly the activity of these two enzymes back to control values, this was without effect on muscle total ceramide or ceramide species contents. If some reports indicated increased muscle ceramide content after HF diet intake( Reference Blachnio-Zabielska, Baranowski and Zabielski 45 , Reference Bruce, Risis and Babb 46 ), other reports failed to show such an increase( Reference Jung and Kang 47 ). This may be due to the type of dietary lipids, the experiment duration and the muscle type( Reference Blachnio-Zabielska, Baranowski and Zabielski 45 ).
SM signalling pathway in muscle is an important factor determining the development of IR( Reference Straczkowski and Kowalska 48 ) and an important direct precursor for ceramide under the action of many different SMase( Reference Khavandgar and Murshed 49 ). In this study, we have found that the HF intake increased muscle SM level, whereas MitoQ intake returned it to the control level. Indeed, the higher enzymatic activity of nSMase by MitoQ may be responsible for this SM decrease. SM synthase was not measurable to better determine the origin of SM increase in the HF group.
It is generally accepted that the type of dietary fat plays a far more significant role in health and disease than its absolute amount( Reference Moussavi, Gavino and Receveur 50 ). The Western diet, characterised by excessive amounts of SFA, n-6 PUFA and trans fatty acids with decreased intake of n-3 PUFA, has been implicated in chronic diseases such as obesity, diabetes and CVD. In parallel, epidemiological, human and animal studies suggest that increased dietary n-3 PUFA may decrease the risk factors for the metabolic syndrome including central adiposity, hyperglycaemia, IR and dyslipidaemia( Reference Poudyal, Panchal and Diwan 51 ). Indeed, although the exact mechanisms of action of n-3 PUFA remain unclear, many possible effects of n-3 PUFA on muscle metabolism related to glucose tolerance and insulin sensitivity have been proposed( Reference Pinel, Morio-Liondore and Capel 52 ). These include potent anti-inflammatory actions mediated directly by their metabolites such as resolvins and protectins( Reference Calder 53 ) and indirectly via the activation of their functional receptor GPR120 that blocks the major NF-κB inflammation pathway( Reference Oh, Talukdar and Bae 54 ). The inhibition of inflammatory cytokine production by n-3 PUFA decreases in turn sphingolipid synthesis, particularly ceramide and SM( Reference Holland, Knotts and Chavez 55 ). Moreover, n-3 PUFA may induce a modification in fatty acid composition of membrane phospholipids, associated with the production of structurally different DAG that may participate in some of the protective effects of n-3 PUFA against various chronic diseases, including IR and diabetes type II( Reference Jude, Martel and Vincent 56 ). In line with this, Lanza et al.( Reference Lanza, Blachnio-Zabielska and Johnson 57 ) have shown that n-3 PUFA protect glucose tolerance, in part by preventing the accumulation of bioactive lipid mediators that interfere with insulin action. The results of the present study show that the HF diet decreased the muscle n-3 PUFA content, and the intake of the mitochondrial-targeted antioxidant MitoQ reestablished it to the control level. This effect may be in part responsible for the favourable effects of MitoQ on body glucose tolerance and insulin sensibility.
In conclusion, the HFD used in this study for 8 weeks was effective at inducing the major features of the metabolic syndrome in our rats – namely, moderate obesity, glucose intolerance associated with muscle mitochondrial dysfunction and marked alteration in lipid composition. Indeed, important modifications in the muscle PL classes (increase in SM and in the SM:PC ratio) in fatty acid composition of total muscle lipids and those of muscle PL (increase in MUFA, particularly C18 : 1 n-9, and decrease in PUFA, particularly PUFA n-3) were observed. In addition, these lipid modifications were accompanied by a significant decrease in mitochondrial respiration. All these modifications could contribute to the development of the obesity, glucose intolerance and IR observed in these rats. Interestingly, MitoQ intake corrected the lipid alterations (decreased muscle TAG, SM and C18 : 1 n-9 and increased PUFA n-3 levels) and restored mitochondrial respiration. Collectively, these results suggest that MitoQ protected rats exposed to a HFD from some metabolic syndrome features probably through lipid metabolism modulation and mitochondrial activity improvement. These findings suggest that MitoQ and related mitochondrial-targeted ubiquinones are promising candidate compounds for future clinical studies in preventing the metabolic syndrome.
Acknowledgements
The authors thank Eva Tolika, Beatrice Bonafos, Beatrice Chabi for their appreciable technical help and Marie-Agnès Chauvin for completion of OxyBlot and OXPHOS Western blot. The authors also thank the staff of the animal facilities for animal use.
The support from the side of the EU project CM0603 is also kindly acknowledged. The authors acknowledge the financial support of the French Lipid Nutrition Group and the National (French) Institute for Agronomic Research and in particular the Human Nutrition Department (Alim-H department).
Author contributions were as follows: study design (C. F.-C. and C. C.), data collection (G. F., K. L., J. R., A. B.-Z., R. E. E., C. F., E. B., J. L.), statistical analysis (C. C. and C. F.-C.), data interpretation (C. F.-C., C. C.), manuscript preparation (C. F.-C., M. P. M., C. C.), literature search (C. F.-C. and C. C.), funds collection (C. F.-C. and C. C.). All the authors have read and approved the final version of this manuscript. M. P. M. who holds patents related to mitochondria-targeted antioxidants as therapies and serves on the scientific advisory board of Antipodean Pharmaceuticals Inc. that develops and commercialises MitoQ.
The authors declare that they have no competing interests, except M. P. M.










