


The prevalence of both obesity and type 2 diabetes (T2D) has increased dramatically in recent decades worldwide. Both conditions represent substantial risk factors for the development of atherosclerotic disease and the resulting increased incidence of myocardial infarction and stroke( Reference Grundy, Pasternak and Greenland 1 ). Atherosclerosis is a chronic inflammatory disease of the large arteries that involves in its earliest stages a non-resolving pro-inflammatory activation of the vascular endothelium, leading to platelet adherence, and activation, adhesion and trans-endothelial migration of monocyte subsets, which then differentiate into macrophages within the developing neointima( Reference Libby, Lichtman and Hansson 2 ).
The development of vascular inflammation and oxidative stress are two key mechanisms involved in endothelial dysfunction and atherosclerosis progression in obesity and T2D and both are promoted independently by hyperglycaemia and insulin resistance (IR)( Reference Hartge, Unger and Kintscher 3 , Reference Mazzone, Chait and Plutzky 4 ). High glucose concentrations, chronic hyperinsulinaemia and obesity, all promote pro-inflammatory gene expression( Reference Hotamisligil, Peraldi and Budavari 5 , Reference Bastard, Maachi and Lagathu 6 ) and endothelial NO synthase uncoupling in endothelial cells, resulting in increased reactive oxygen species (ROS), reduced NO and a reduction in endothelial-dependent relaxation and associated vascular changes( Reference Yan, Schmidt and Anderson 7 – Reference Son 9 ).
Extracellular vesicles are small ( < 1 μm) circulating vesicles that can be classified as smaller exosomes (80–100 nm), released by an endosomal pathway( Reference van der Pol, Böing and Harrison 10 – Reference Théry, Ostrowski and Segura 12 ) or larger, more heterogeneous microvesicles (MV; 100–1000 nm). These are released from cells including endothelial cells, leucocytes, platelets and erythrocytes by blebbing of the plasma membrane during activation and apoptosis( Reference van der Pol, Böing and Harrison 10 – Reference Théry, Ostrowski and Segura 12 ). Plasma MV are increased during disease processes, including cancer, CVD and autoimmunity (for reviews see Diamant et al. ( Reference Diamant, Nieuwland and Pablo 15 ), Tramontano et al. ( Reference Tramontano, Lyubarova and Tsiakos 16 ), Chen et al. ( Reference Chen, Feng and Li 17 )). MV can be identified by the presence of exposed phosphatidyl serine on their plasma membranes and by the presence of surface markers referable to their parent cells. They can bind to cells using membrane receptors and contain ‘cargo’ such as proteins, mRNA and microRNA, which they can deliver to recipient cells and thus activate intracellular signalling pathways and gene transcription. Thus, MV may be useful plasma biomarkers as well as contributing to disease processes by altering signal transduction( Reference Théry, Ostrowski and Segura 12 ).
Chronic inflammation of the endothelium may be one of the triggers for MV release from circulating cells. Our previous work demonstrated release of pro-oxidant, pro-coagulant leucocyte, platelet and erythrocyte-derived MV in whole blood flowing over inflamed endothelium that could induce ROS formation in quiescent endothelium( Reference Macey, Wolf and Lawson 13 , Reference Holtom, Usherwood and Macey 14 ). Increased numbers of MV have been identified in the plasma of patients with elevated plasma LDL-cholesterol, obesity, metabolic syndrome and T2D, all of which represent risk factors for CVD( Reference Diamant, Nieuwland and Pablo 15 – Reference Chen, Feng and Li 17 ).
The present study aims to determine whether a well-described rodent model of obesity and IR, which has previously been shown to have a pro-inflammatory phenotype( Reference Sena, Matafome and Louro 18 ), also leads to production of increased numbers of circulating pro-inflammatory MV. We used plasma obtained from rats that had been fed a high-fat diet (HFD) to identify the cellular origin of the MV released during obesity and determined whether the purified MV could directly induce pro-inflammatory changes in rat cardiac endothelial cells (RCEC).
Methods
Animal model
The present study was approved by the Royal Veterinary College's Welfare and Ethics Committee and was carried out under UK Home Office licenses to comply with the Animals (Scientific Procedures) Act 1986. Six-week-old male Wistar rats (Charles River) were maintained at 22 ± 0·5°C under a 12 h light–12 h dark cycle. Ten were fed a standard maintenance chow diet (Special Diet Services) and ten were fed a HFD, providing 60 % energy as fat (RD12492; Research Diets) ad libitum for 20 weeks. An intraperitoneal insulin tolerance test was conducted in overnight fasted rats to confirm the development of IR. Briefly, blood glucose was measured immediately prior (time = 0) to an intraperitoneal injection of 3 nmol/kg (0·5 IU/kg) insulin (Actrapid; Novo Nordisk), and 15, 30, 60 and 90 min afterwards using an Accuchek Aviva handheld glucometer (Roche). Plasma insulin concentration was measured using a rat insulin RIA kit (Millipore) at time 0. Body mass and epididymal fat pad mass were recorded at euthanasia.
Blood was collected by cardiac puncture under terminal anaesthesia with pentobarbitone and 1 ml was collected into tubes containing 100 μl 3·2 % sodium citrate, followed by centrifugation at 2000 g for 1 min to separate plasma. Plasma was collected, stored at − 80°C and thawed on ice before use.
Materials
Phycoerythrin (PE)-IgGI k isotype control and fluorescein isothiocyanate (FITC)-IgGI k isotype control, PE anti-CD31 (PECAM-1; used as endothelial marker) and PE anti-α/β T-cell receptor (TCR; used as T cell marker) were from BD Bioscience. PE anti-CD106 (vascular cell adhesion molecule-1; VCAM-1; expressed by activated endothelial cells) was from BioLegend. PE Cy7 annexin V (AnnV; to detect exposed phosphatidyl serine), PE anti-CD45.2 (pan-leucocyte marker) and PE anti- CD61 (integrin β3; platelet marker) were from eBioscience. Fluorescein isothiocyanate anti-CD43 (pan-leucocyte marker apart from B Cells), PE anti-CD11b (activated leucocyte marker) and fluorescein isothiocyanate anti-macrophage antibody were from AbDSerotec.
Latex sizing beads (1·1 μm) and Dihydrorhodamine 123 were from Sigma and enumeration beads from Bangs Laboratories (supplied by Stratech Scientific). Tissue culture medium was from PAA or Sigma. Full endothelial cell medium (EC medium) contained HEPES-buffered M199 supplemented with penicillin/streptomycin, 2 mm-l-glutamine, 20 % fetal calf serum, 0·55 mg/ml heparin and 275 μg/ml endothelial cell growth supplement. 1 × Tyrode's buffer was prepared daily (139 mm-NaCl, 3 mm-KCl, 17 mm-NaHCO3, 3 mm-CaCl2, 1 mm-MgCl2). Annexin V Binding buffer was from eBioscience. All buffers were passed through 0·22 μm syringe filters before use.
Enumeration and phenotyping of microvesicle populations by flow cytometry
Flow cytometric analysis of the same volume of antibody-labelled plasma from each animal was carried out as described previously( Reference Macey, Wolf and Lawson 13 , Reference Holtom, Usherwood and Macey 14 ), using a FACS Canto II (BD Biosciences), utilising three lasers, eight colours, a standard optical filter setup and FACS Diva Software version 6.1 (BD Biosciences). The instrument was calibrated daily using Cell Tracker Beads (BD Biosciences). MV were identified by their characteristic forward (FSC-A; indicates size of particles) and side scatter (SSC-A; indicates granularity of particles) and their ability to bind to cell-specific monoclonal antibodies. Plasma samples were diluted 1:10 in Tyrode's buffer and 50 μl diluted plasma was incubated on ice with antibodies or isotype controls. After 15 min, 450 μl of Tyrode's or Annexin V binding buffer was added. 1·09 μm latex beads (Sigma) were added to each sample to allow detection of particles < 1 μm. 10 μl Absolute Count beads (Bang's Laboratories) were added to each sample to allow internal calibration of the number of events < 1·1 μm according to the following equation. Samples were acquired for 2 min on a low flow rate (online Supplementary Fig. S1). Isotype controls were used to adjust voltages for fluorescein isothiocyanate, PE or Cy-7 for each sample. Negative gates were set to 1 % of total counts for fluorescence. A platelet-gate was additionally plotted on the FSC/SSC plots according to the characteristic ‘cloud’ of light-scatter characteristic of platelet populations (online Supplementary Fig. S1(C)( Reference Macey, Wolf and Lawson 13 )). For each phenotypic marker, MV were counted by reference to enumeration beads:
 $$\begin{eqnarray} MV\,density/\mu l = (number\,of\,enumeration\,beads\,added/\mu l\times events\,counted\times dilution\,factor)/(number\,of\,enumeration\,beads\,counted). \end{eqnarray}$$
$$\begin{eqnarray} MV\,density/\mu l = (number\,of\,enumeration\,beads\,added/\mu l\times events\,counted\times dilution\,factor)/(number\,of\,enumeration\,beads\,counted). \end{eqnarray}$$
Measurement of microvesicle-induced reactive oxygen species in rat endothelial cells using dihydrorhodamine 123
We conducted a small pilot study to determine a suitable number of pro-inflammatory MV to add using THP-1 monocytes (human monocyte cell line TIB202 available from the American Type Culture Collection) that had been differentiated with phorbol myristate acetate (PMA) 200 ng/ml for 72 h. Briefly, differentiated THP-1 (1 × 106 per well) were washed to remove non-adherent cells, and treated with 10 μg/ml lipopolysaccharide (LPS) for 24 h (medium was removed, cells washed and replaced with fresh medium after 8 h to remove traces of unbound LPS). MV were analysed by flow cytometry with enumeration beads as described in Materials and Methods. Appropriate volumes of conditioned medium from untreated or LPS-treated THP-1 macrophages equating to final concentrations of 1000, 200, 100, 50 or 10 MV/μl were centrifuged at 17 000 g for 15 min to pellet MV and re-suspended in 500 μl M199/10 % fetal calf serum. Conditioned medium (100 μl) was added to human umbilical vein endothelial cells seeded onto white opaque ninety-six well plates (2 × 104 per well) and pre-loaded with 5 μm dihydrorhodamine 123, followed by analysis on a fluorescent plate reader (Wallac Victor 2 1420) after 24 h. LPS (10 mg/ml) was added to human umbilical vein endothelial cells as a positive control (online Supplementary Fig. S3). From this we determined that a minimum of 100 MV/μl of MV released from inflamed cells was sufficient for eliciting ROS production in EC. In contrast, up to 1000 MV/μl of MV from quiescent cells had no effect on ROS.
Primary cultures of RCEC isolated from the PVG.AO-RT1μ strain were obtained from Professor Marlene Rose (Imperial College, London) at no cost. RCEC were cultured in EC medium and seeded at 25 000 cells per well on white opaque ninety-six well plates and allowed to adhere overnight. The culture medium was replaced with EC medium containing 1μm-dihydrorhodamine 123, followed by 10-min incubation in the dark. Meanwhile, MV were isolated by centrifugation of 50 μl of each plasma sample at 17 000 g for 15 min at 4°C. Pellets were re-suspended in 500 μl EC medium (a one in ten dilution from the original plasma; approximately 900 MV/μl from HFD-fed rats was added to the cells) and added in triplicate. Fluorescence was read at 0, 5, 10, 15, 30 min, 1, 2, 4 and 24 h on a fluorescent plate reader (Wallac Victor 2 1420 Multilabel counter; Perkin Elmer). Positive (1 nm-H2O2) and negative (100 μl of medium) controls were also analysed (n 4 RCEC isolates).
Measurement of CD106 (vascular cell adhesion molecule-1) expression in microvesicle-treated rat cardiac endothelial cells
RCEC were seeded at 200 000 cells per well. The following day MV were isolated by centrifugation of 50 μl (approximately 50 000 MV in total) of each plasma sample at 17 000 g for 10 min at 4°C. The supernatant was removed and the pellet (containing MV) was re-suspended in 500 μl EC medium (a one in ten dilution from the original plasma–approximately 900 MV/μl from HFD-fed rats). Medium was removed from adhered RCEC, replaced with MV-containing medium and the cells were incubated overnight at 37°C. Negative (medium alone) and positive (LPS 100 ng/ml) controls were included. RCEC were disaggregated with 1 × trypsin/EDTA for 5 min. Non-adherent cells were pelleted and incubated with anti-CD106 or isotype control antibodies on ice for 20 min, then FACS FIX (0·5 % formaldehyde in PBS) was added to each tube and samples were immediately analysed. Median fluorescence intensity for each sample was recorded (n 3 RCEC isolates).
Statistical analysis
Data analysis was carried out using GraphPad Prism version 6 (GraphPad Software). All data are means with their standard errors. A D'Agostino and Pearson omnibus normality test was carried out on all MV data and consequently all were analysed using Mann–Whitney U or Kruskal–Wallis tests followed by Dunn's multiple comparison test as appropriate. For differences between the treatment groups across time points, a two-way ANOVA was used, accompanied by the Tukey post hoc test where appropriate. Correlation measurements were carried out using Spearman's correlation for non-parametric data (r 2). Student's t tests were used to compare total rat and fat depot masses and plasma insulin and two-way repeated measures ANOVA for intraperitoneal insulin tolerance test data. P< 0·05 was accepted as significant.
Results
High-fat diet feeding-induced obesity is associated with increased phosphatidyl serine-positive microvesicle release
Feeding of a HFD for 20 weeks resulted in increased body mass (746 (sem 20) v. 523 (sem 30) g; P< 0·001) and epididymal fat pad mass as a percentage of body mass (3·11 (sem 0·17) v. 1·44 (sem 0·13) %: P< 0·001) v. chow-fed rats, consistent with the development of obesity. Fasting plasma insulin was also increased in HFD- v. chow-fed rats (5·84 (sem 0·46) v. 2·44 (sem 0·35) ng/ml; P< 0·001), while blood glucose concentrations were suppressed less effectively in HFD-fed rats during an intraperitoneal insulin tolerance test (online Supplementary Fig. S2), confirming the development of IR. As shown in online Supplementary Fig. S1 and Fig. 1(a), there were 12·7-fold more MV identified in the plasma of HFD-fed rats compared with that in the plasma of chow-fed rats (P< 0·001). There was also an 8-fold higher Annexin V binding to MV in HFD plasma than in chow plasma (P< 0·0001; Fig. 1(b)), indicative of increased phosphatidyl serine exposure and loss of membrane asymmetry.
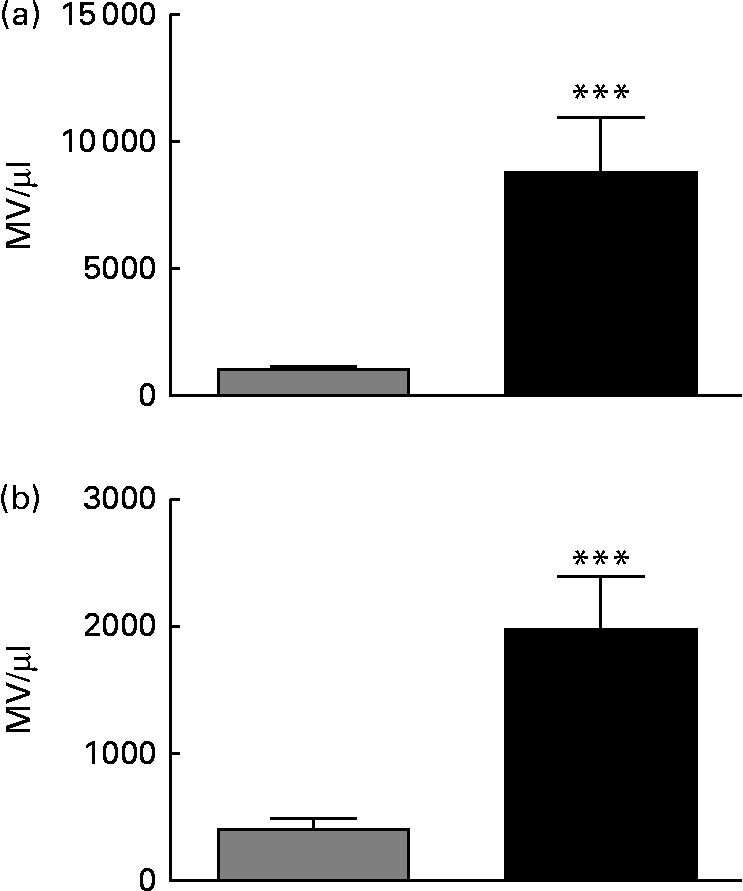
Fig. 1 Annexin V+ microvesicles (MV) are elevated in the plasma of rats fed a high-fat diet (HFD) for 20 weeks. MV in plasma samples from chow-fed (![]() ) or HFD-fed (■) rats were enumerated by flow cytometry. (a) Total MV/μl counted in each plasma sample using the gating strategy outlined in Methods and illustrated in online Supplementary Fig. S1(A). (b) Annexin V+ MV. n 9–10 rats/group. Values are means, with their standard errors represented by vertical bars. *** Mean value was significantly different from that of the chow-fed group (P< 0·001).
) or HFD-fed (■) rats were enumerated by flow cytometry. (a) Total MV/μl counted in each plasma sample using the gating strategy outlined in Methods and illustrated in online Supplementary Fig. S1(A). (b) Annexin V+ MV. n 9–10 rats/group. Values are means, with their standard errors represented by vertical bars. *** Mean value was significantly different from that of the chow-fed group (P< 0·001).
Increased numbers of CD45+, CD43+ or TCR+ microvesicles are released in rat obesity
The cellular sources of MV identified in Fig. 1 were characterised using fluorescently conjugated antibodies directed against CD43, CD45 or TCR, proteins expressed on the surface of leucocytes (Fig. 2). CD45+ MV (pan-leucocyte marker) were 7·5-fold increased after HFD feeding (P= 0·0029; Fig. 2(a)). Furthermore, MV expressing CD43, a marker of T-lymphocytes and monocytes (2·5-fold, P= 0·0354; Fig. 2(b)), the antigen recognised by MAC387 (a monocyte marker; 22-fold, P= 0·0018; Fig. 2(c)), αβ TCR (17·3-fold, P< 0·0001; Fig. 2(d)) and CD11b (2·0-fold, P= 0·0147; Fig. 2(e)), were all increased in obese rat plasma, indicating an effect of HFD feeding on all major classes of leucocyte-derived MV, including those released by activated cells. Furthermore, there was a significant correlation between blood glucose level and numbers of MAC387+ MV (r 0·789, P= 0·007) in HFD-fed rats (Fig. 2(f)).

Fig. 2 Leucocyte-derived microvesicles (MV) are increased in the plasma of rats fed a high-fat diet (HFD) for 20 weeks. MV with leucocyte surface markers were enumerated by flow cytometry in plasma samples from chow-fed (![]() ) or HFD fed (■) rats. (a) CD45+ MV (pan-leucocyte marker). (b) CD43+ MV (all leucocytes except for B cells). (c) Monocyte-derived MV (MAC387 antigen+). (d) TCR+ MV (T-cell marker). (e) CD11b+ MV (activated leukocytes). n 9–10 rats/group. Values are means, with their standard errors represented by vertical bars. Mean value was significantly different from that of the chow-fed group: * P< 0·05, ** P< 0·01, **** P< 0·0001. (f) Correlation of MAC387+ MV with glucose (mm).
) or HFD fed (■) rats. (a) CD45+ MV (pan-leucocyte marker). (b) CD43+ MV (all leucocytes except for B cells). (c) Monocyte-derived MV (MAC387 antigen+). (d) TCR+ MV (T-cell marker). (e) CD11b+ MV (activated leukocytes). n 9–10 rats/group. Values are means, with their standard errors represented by vertical bars. Mean value was significantly different from that of the chow-fed group: * P< 0·05, ** P< 0·01, **** P< 0·0001. (f) Correlation of MAC387+ MV with glucose (mm). ![]() , MAC387+ MV HFD;
, MAC387+ MV HFD; ![]() , MAC387+ MV chow. R 0·089 P= 0·007.
, MAC387+ MV chow. R 0·089 P= 0·007.
Platelet and endothelial cell-derived microvesicles are increased in plasma from obese rats
As shown in Fig. 3, plasma from HFD-fed rats contained 9·4-fold more endothelial cell-derived CD31+MV/μl, compared to chow-fed rats (P= 0·0185; Fig. 3(a)). In addition, VCAM-1+MV, derived from activated EC, tended to be increased in HFD- v. chow-fed rats (by 73 %, P= 0·088; Fig. 3(b)). Surprisingly, Annexin V+ platelet-derived MV (CD61+) appeared to be very rare in plasma from chow-fed rats (4 (sem 1) MV/μl), but were markedly more abundant in HFD plasma (1055 (sem 609) MV/μl, P= 0·0039), therefore we reanalysed the data to include both Ann V+ and Ann V– CD61+ MV (174 (sem 105) in chow-fed v. 1570 (sem 1277) in HFD-fed plasma P= 0·0281; Fig. 3(c)). To confirm that platelet-derived MV were increased, the FSC/SSC plots were re-analysed to determine the number of CD61+ (AnnV+ and Ann V−) MV in the classic platelet cloud (129 (sem 51·0) CD61+ MV/μl in chow- and 349 (sem 105) in HFD-fed rat plasma; P= 0·0039; online Supplementary Fig. S1(C), Fig. 3(d)). There were significant correlations between blood glucose level and numbers of both CD31+ MV (r 2 0·645, P= 0·044) and CD61+ MV (r 2 0·541, P= 0·0153) in HFD-fed rats (Fig. 3(e) and (f)).

Fig. 3 CD31+ and CD61+ microvesicles (MV) are raised in the plasma of rats fed a high-fat diet (HFD) for 20 weeks. CD31+ (endothelial) MV (a, b) or CD61+(platelet) (c, d) surface markers were enumerated by flow cytometry in plasma samples from chow-fed (![]() ) or HFD-fed (■) rats. (a) CD31+ MV (endothelial cell marker). (b) CD106+ MV (activated endothelial cells). (c) CD61+ MV (platelets). (D) CD61+ MV in platelet gate illustrated in online Supplementary Fig. S1(C). n 9–10 rats/group. Values are means, with their standard errors represented by vertical bars. Mean value was significantly different from that of the chow-fed group: * P< 0·05, ** P< 0·01. † Mean value was marginally significantly different from that of the chow-fed group (P< 0·1). (e) Correlation of CD31+ MV in HFD plasma and glucose R 0·645 P= 0·044.
) or HFD-fed (■) rats. (a) CD31+ MV (endothelial cell marker). (b) CD106+ MV (activated endothelial cells). (c) CD61+ MV (platelets). (D) CD61+ MV in platelet gate illustrated in online Supplementary Fig. S1(C). n 9–10 rats/group. Values are means, with their standard errors represented by vertical bars. Mean value was significantly different from that of the chow-fed group: * P< 0·05, ** P< 0·01. † Mean value was marginally significantly different from that of the chow-fed group (P< 0·1). (e) Correlation of CD31+ MV in HFD plasma and glucose R 0·645 P= 0·044. ![]() , CD31+ MV HFD;
, CD31+ MV HFD; ![]() , CD31+ MV chow. (f) Correlation of CD61+ MV in HFD plasma with glucose (mm).
, CD31+ MV chow. (f) Correlation of CD61+ MV in HFD plasma with glucose (mm). ![]() , CD61+ MV chow;
, CD61+ MV chow; ![]() , CD61+ MV HFD. R
2 0·541, P= 0·0153.
, CD61+ MV HFD. R
2 0·541, P= 0·0153.
Exposure of quiescent endothelial cells to microvesicles derived from obese rats induces vascular cell adhesion molecule-1 expression
VCAM-1 (CD106) is expressed by cytokine-activated endothelium and is a marker of inflammation( Reference Libby, Lichtman and Hansson 2 ). As shown in Fig. 4, incubation of RCEC with MV derived from plasma of HFD-fed rats but not from chow-fed rats induced an increase in VCAM-1 median fluorescence intensity on the cell surface to a similar extent to that induced by positive control LPS treatment (VCAM-1 increased by 59 % in HFD v. chow endothelium; P= 0·0275).

Fig. 4 CD106 (vascular cell adhesion molecule-1 (VCAM-1)) expression is increased on endothelial cells treated with microvesicles (MV) from high-fat diet (HFD) fed rats. MV from chow-fed rats (![]() ) or HFD-fed rats (■) for 20 weeks were isolated from plasma by centrifugation at 17 000
g
and used to stimulate primary cultures of rat cardiac endothelial cells for 18 h. VCAM-1 mean fluorescence intensity (MFI) on rat cardiac endothelial cells surface was analysed by flow cytometry. 10 ng/ml lipopolysaccharide (LPS) used as a positive control (hatched), n 3 RCEC isolates. Values are means, with their standard errors represented by vertical bars. * Mean value was significantly different from that of the chow-fed group (P< 0·05). a.u., Arbitrary units.
) or HFD-fed rats (■) for 20 weeks were isolated from plasma by centrifugation at 17 000
g
and used to stimulate primary cultures of rat cardiac endothelial cells for 18 h. VCAM-1 mean fluorescence intensity (MFI) on rat cardiac endothelial cells surface was analysed by flow cytometry. 10 ng/ml lipopolysaccharide (LPS) used as a positive control (hatched), n 3 RCEC isolates. Values are means, with their standard errors represented by vertical bars. * Mean value was significantly different from that of the chow-fed group (P< 0·05). a.u., Arbitrary units.
Exposure of quiescent endothelial cells to microvesicles derived from obese rats induces reactive oxygen species formation
ROS formation was measured over 24 h using dihydrorhodamine 123. At all time points H2O2 treatment led to increased ROS formation v. either control or addition of MV, as expected (Fig. 5). However, after 24-h treatment, ROS production was increased in HCEC treated with MV from HFD-fed rats compared with the untreated control or chow MV (30 % increase in HFD v. chow; P< 0·0001).

Fig. 5 Reactive oxygen species (ROS) induction in rat endothelial cells by microvesicles (MV) from chow- or high-fat diet (HFD)-fed rats. MV were isolated from plasma by centrifugation at 17 000
g
and used to stimulate primary cultures of rat cardiac endothelial cells that had been loaded with dihydrorhodamine 123. MV from rats fed chow (![]() ) or HFD (
) or HFD (![]() ) for 20 weeks. 1 nm-hydrogen peroxide used as a positive control (
) for 20 weeks. 1 nm-hydrogen peroxide used as a positive control (![]() ), untreated (
), untreated (![]() ); n 4 RCEC isolates. Two-way non-parametric ANOVA followed by Tukey. Values are means, with their standard errors represented by vertical bars. **** Mean value was significantly different from that of the chow-fed group at 24 h after treatment (P< 0·0001). ‡‡‡‡ Mean values for the hydrogen peroxide-treated group were significantly higher than for all other groups at all time points (P< 0·0001).
); n 4 RCEC isolates. Two-way non-parametric ANOVA followed by Tukey. Values are means, with their standard errors represented by vertical bars. **** Mean value was significantly different from that of the chow-fed group at 24 h after treatment (P< 0·0001). ‡‡‡‡ Mean values for the hydrogen peroxide-treated group were significantly higher than for all other groups at all time points (P< 0·0001).
Discussion
There is a growing literature demonstrating that chronic diseases with an inflammatory component are characterised by increased release of MV into plasma. In the present study, we have used a well-characterised rodent model to show that obesity is associated with substantially elevated numbers of circulating MV, in agreement with data from recent studies of human obesity and diabetes( Reference Diamant, Nieuwland and Pablo 15 – Reference Chen, Feng and Li 17 , Reference Stepanian, Bourguignat and Hennou 19 – Reference Zhang, McGeoch and Johnstone 21 ). In addition, phenotypic analysis of these MV revealed that they were derived from endothelial cells (CD31+), platelets (CD61+), T cells (TCRα/β) and monocytes (MAC387+) – key cell populations involved in the initiation and progression of atherosclerosis whose numbers are also elevated in human cardio-metabolic disease( Reference Chen, Feng and Li 17 , Reference Feng, Chen and Luo 22 , Reference Kranendonk, de Kleijn and Kalkhoven 23 ). Furthermore, the numbers of MV correlated with the glucose levels in these animals, again consistent with human studies( Reference Kranendonk, de Kleijn and Kalkhoven 23 ). Recent studies (e.g. do Carmo et al. ( Reference do Carmo, Rogero and Paredes-Gamero 24 )) suggest that long-term feeding of a HFD leads to bone marrow hyperplasia and modulation of haematopoiesis, which in turn leads to higher numbers of circulating leucocytes, which could at least in part account for the observed increase in circulating leucocyte MV in the HFD-fed rats. Interestingly, we found a large proportion of the CD61+ MV did not bind Annexin V, suggesting that HFD led to an increase in platelet MV release due to increased turnover as has been described in human samples( Reference Connor, Exner and Ma 25 ) rather than activation and release of procoagulant MV.
The overall numbers of platelet-derived MV detected are in agreement with another recent study carried out in streptozotocin-treated rats( Reference Hosseinzadeh, Zahmatkesh and Zarrindast 26 ). Interestingly, another study that subjected younger rats (13 weeks old rather than 26 weeks old) to hypoxia described much higher numbers of platelet-derived MV, both in control and in hypoxia-exposed animals( Reference Tual-Chalot, Guibert and Muller 27 ). This may reflect an age-dependent decline in the release of MV from platelets in the rat, although this possibility awaits confirmation.
MV were able to elicit pro-inflammatory effects in quiescent endothelial cells, a finding consistent with the existing theory that circulating MV do not merely represent a mechanism to remove cellular debris from dying or activated cells, but have functional importance in transcellular communication during the progression of chronic inflammatory diseases, and may be important for increasing endothelial dysfunction in plaque-prone areas, a pre-requisite for atheroma formation( Reference van der Pol, Böing and Harrison 10 ). The present work also highlights the usefulness of this rodent model to further study the influence of diet on MV release and the effects of MV on vascular inflammation. To our knowledge, this is the first study to demonstrate the potential functional importance of elevated numbers of MV observed in obesity for inciting endothelial inflammatory responses. However, although we have shown a relationship between plasma glucose and numbers of various MV subsets, it is unclear as yet which of the obesity-associated metabolic, endocrine or pro-inflammatory changes may be responsible for the increased MV generation.
MV derived from cells of monocyte/macrophage lineage, T cells, endothelial cells and platelets have been identified in human atherosclerotic lesions( Reference Canault, Leroyer and Peiretti 28 ) and shown to contribute to plaque instability, in part by promoting monocyte adhesion and trans-endothelial migration via enhanced surface expression of endothelial intercellular adhesion molecule-1( Reference Rautou, Leroyer and Ramkhelawon 29 ). In the present study, MV derived from HFD-fed rats induced VCAM-1 expression, which is another key adhesion molecule involved in firm adhesion and trans-endothelial migration of monocytes in rodent models of atherosclerosis( Reference Iiyama, Hajra and Iiyama 30 ). Further work is required to determine the full extent of pro-inflammatory gene expression up-regulated by MV derived from HFD- v. chow-fed rats.
It is likely that the individual species of MV present in the mixed populations isolated from patients or animal models have disparate effects, but together have a net pro- or anti-atherogenic effect on endothelia. Further work is required to dissect the responses elicited by individual MV species in order to fully understand how they contribute to vascular inflammation and atherosclerosis. However, it is still not technically possible to satisfactorily separate in vivo generated MV to determine their function. We have previously developed an in vitro flow system to model interactions between blood and inflamed endothelium under haemodynamic flow conditions and showed that this leads to enhanced release of MV from leucocytes, platelets and erythrocytes. Furthermore, these MV were able to induce ROS in quiescent endothelium, suggesting that they are similar to in vivo generated MV( Reference Macey, Wolf and Lawson 13 , Reference Holtom, Usherwood and Macey 14 ). Using isolated blood cells from animals fed a defined HFD or from T2D patients, and autologous isolated endothelial cells (rodent) or explanted blood vessels (from diabetic peripheral arterial disease amputees) it should in future be possible to determine more accurately the role of individual species of MV in endothelial dysfunction.
One of the limitations to the present study is that only one subset of extracellular vesicles was measured, namely, the MV population >200 nm in diameter, due to difficulties distinguishing smaller particles from background noise by conventional flow cytometry. It is likely that in addition to release of the larger MV there is also increased production of pro-inflammatory exosomes and smaller ( < 200 nm) MV, in development of obesity and IR in this rodent model in agreement with human studies( Reference Ferrante, Nadler and Pillai 31 ). A number of methods for exosome measurement and isolation have been described recently, including Nanotracking analysis and dynamic light scattering (for review see Yellon & Davidson( Reference Yellon and Davidson 32 ), Mulcahy et al. ( Reference Mulcahy, Pink and Carter 33 )). However, at the present time, it is not possible to identify the cellular sources of exosomes, and nanotracking analysis and DLA do not easily enable identification of the cellular source of MV, thus flow cytometric analysis is currently the most established technology for immune-phenotyping of MV released by inflamed and activated cells. In addition, we utilised frozen and thawed plasma samples. Recent studies by several groups have recommended analysis of fresh plasma or whole blood for extracellular vesicle analysis; however, use of citrate as anti-coagulant followed by freezing of plasma at − 80°C has been shown to be an acceptable alternative (for recent reviews see Lacroix et al. ( Reference Lacroix, Judicone and Mooberry 34 ), van der Meel et al. ( Reference van der Meel, Krawczyk-Durka and van Solinge 35 )). We used MV from a similar volume of plasma from HFD- or chow-fed rats for our functional studies. This was based on pilot studies with MV derived from single (human) cell types in tissue culture experiments, which suggested that the number of MV added is not a significant factor for their pro-inflammatory or other effects (online Supplementary Fig. S3), but instead the stimulus that has been used to generate the MV is more important. A similar approach has been taken in other studies where numbers of MV approximating to those found circulating have been added( Reference Tual-Chalot, Guibert and Muller 27 ).
Obesity and IR induce oxidative stress and inflammation in the endothelium, features of endothelial dysfunction that predispose to atherosclerotic lesion formation( Reference Hartge, Unger and Kintscher 3 , Reference Mazzone, Chait and Plutzky 4 ). Furthermore, endothelial cells from T2D patients display defective insulin signalling, increased nitrotyrosine, ROS and increased NF-κB p65 and intercellular adhesion molecule-1 expression( Reference Tabit, Shenouda and Holbrook 36 ). It is possible that increased VCAM-1 expression is mediated by the observed ROS production, which might be downstream of RAC activation (for review see Marcos-Ramiro et al. ( Reference Marcos-Ramiro, García-Weber and Millán 37 )). Although limitations in the plasma sample volume collected from each animal prevented us from investigating this mechanism during the present study, it could be investigated using ROS scavengers or dominant negative RAC constructs in in vitro experiments. It is likely that these effects are also mediated at least in part by chronic activation of protein kinase C, which has been described in micro- and macro-vascular tissue and circulating leucocytes in human T2D and rodent models( Reference Tabit, Shenouda and Holbrook 36 ).
In agreement with this, protein kinase C is activated during culture of rat aortic endothelial cells, smooth muscle cells and circulating monocytes in hyperglycaemic conditions( Reference Das Evcimen and King 38 ). Protein kinase C activation is associated with phosphatidyl serine externalisation and CD43 expression on MV in neutrophils( Reference Nusbaum, Lainé and Bouaouina 39 ), suggesting a direct link between IR and increased levels of circulating MV. Endothelial and platelet-derived MV identified in the plasma of patients with metabolic syndrome have also been shown to have a pro-oxidant effect on endothelium, inducing NO and ROS production( Reference Agouni, Ducluzeau and Benameur 40 ).
Hyperglycaemia and impaired insulin signalling leading to endothelial inflammation and protein kinase C activation could contribute to the increased numbers of circulating MV observed in obese rats and human subjects. Moreover, given that oxidative stress and pro-inflammatory gene expression are key characteristics of diabetic endothelial dysfunction( Reference Hartge, Unger and Kintscher 3 , Reference Mazzone, Chait and Plutzky 4 ) and our findings that MV from HFD-fed rats lead to increases in both ROS and VCAM-1 expression, we and others have proposed that MV released from multiple cell types under diabetogenic conditions are functional particles that contribute to systemic vascular inflammation and endothelial dysfunction, leading to macrovascular changes and potentially the accelerated atherosclerotic lesion progression that is observed in T2D( Reference Chen, Feng and Li 17 , Reference Feng, Chen and Luo 22 , Reference Kranendonk, de Kleijn and Kalkhoven 23 ). Further work is required to determine the exact mechanisms leading to MV release under diabetogenic conditions, and the rat model used in the present study may prove useful to help determine how MV release and their functional properties can be altered in a physiologically relevant in vivo context, in response to dietary composition or therapeutic substances.
Supplementary material
To view supplementary material for this article, please visit http://dx.doi.org/10.1017/S0007114515001117
Acknowledgements
D. K. A. was funded by a PhD studentship from Diabetes UK. We would like to thank the Royal Veterinary College BSU staff for animal care.
There are no financial or other relationships that might lead to a conflict of interest.









