


Preamble
Inflammation is a normal host defence mechanism that protects the host from infection and other insults; it initiates pathogen killing as well as tissue repair processes and helps to restore homeostasis at infected or damaged sites. It is typed by redness, swelling, heat, pain and loss of function, and involves interactions among many cell types and the production of, and responses to, a number of chemical mediators. Normally, the host is tolerant to microbes and other environmental components that do not pose a threat. This tolerance involves only a limited host response or an active response that is tightly controlled. Where an inflammatory response does occur, it is normally well regulated in order that it does not cause excessive damage to the host, is self-limiting and resolves rapidly. This self-regulation involves the activation of negative feedback mechanisms such as the secretion of anti-inflammatory cytokines, inhibition of pro-inflammatory signalling cascades, shedding of receptors for inflammatory mediators and activation of regulatory cells. As such, and controlled properly, regulated inflammatory responses are essential to remain healthy and maintain homeostasis. Pathological inflammation involves a loss of tolerance and/or of regulatory processes. Where this becomes excessive, irreparable damage to host tissues and disease can occur. Typically, diseases or conditions with a well-recognised inflammatory component are treated with general or specific anti-inflammatory pharmaceuticals. However, since many dietary components may influence various elements of inflammation, nutrition may play a role in predisposing to inflammatory conditions and altered nutrition may be useful in therapy of such conditions. A workshop was held in Oporto, Portugal, on 29–31 May 2006. The workshop aimed to consider the role of inflammation in various diseases and conditions, to identify common and unique mechanisms and markers of inflammation, and to review and consolidate evidence that dietary components can influence inflammatory processes and to understand their mechanisms of action. The present paper is based upon the presentations made at the workshop and the subsequent discussions.
General aspects of the inflammatory process
Inflammation may be classified into four types, the mechanisms of which in part overlap: (i) inflammation caused by innate and acquired immunity against infectious agents, in which cells are activated and mediators released to prevent or combat infection and remove foreign material; (ii) inflammation caused by different inhaled agents (‘irritants’) like diesel exhaust particles, ozone and endotoxin; (iii) allergic inflammation, in which specific IgE antibodies bound to mast cells upon cross-linking by allergen cause the immediate release of a number of inflammatory mediators and activation of inflammatory cells; and (iv) neurogenic inflammation, mediated by the neural system. Common to these forms of inflammation is that they have an afferent phase, in which the presence of a ‘foreign material’ is sensed by some types of cell, and an efferent phase, in which an inflammatory response is generated to eliminate the perceived hostile intruder. The purpose of the inflammatory response to micro-organisms is obvious, and the response is beneficial and necessary to protect the integrity of the body as long as it does not become unnecessarily destructive or long-lasting. Inflammation caused by non-pathogenic agents can also be beneficial and remove the foreign material e.g. by increasing mucous production and increasing the number of phagocytic cells, but it may also have negative health effects in particular if long-lasting. Allergic inflammation is triggered by minute amounts of innocuous foreign material, so-called allergens from plants, insects, animals and foods, and serves no obvious beneficial purpose, except that similar responses may protect against certain infectious agents (parasites). Neurogenic inflammation may be looked upon as an adjuct mechanism to the other three types of inflammation. Irrespective of the cause of the inflammation, the response involves four major events.
(i) An increased blood supply to the site of inflammation.
(ii) Increased capillary permeability caused by retraction of endothelial cells. This permits larger molecules, not normally capable of traversing the endothelium, to do so and thus delivers some soluble mediators to the site of inflammation.
(iii) Leucocyte migration from the capillaries into the surrounding tissue (Fig. 1). This is promoted by release of chemoattractants from the site of inflammation and by the upregulation of adhesion molecules on the endothelium. Once in the tissue the leucocytes move to the site of inflammation.
(iv) Release of mediators from leucocytes at the site of inflammation (Fig. 1). These may include lipid mediators (e.g. PG, leukotrienes), peptide mediators (e.g. cytokines), reactive oxygen species (ROS, e.g. superoxide), amino acid derivatives (e.g. histamine) and enzymes (e.g. matrix proteases) depending upon the cell type involved, the nature of the inflammatory stimulus, the anatomical site involved and the stage during the inflammatory response. These mediators normally would play a role in host defence, but when produced inappropriately or in an unregulated fashion, they can cause damage to host tissues, leading to disease. Several of these mediators may act to amplify the inflammatory process acting, for example, as chemoattractants. Some of the inflammatory mediators may escape the inflammatory site into the circulation and from there they can exert systemic effects. For example, the cytokine IL-6 induces hepatic synthesis of the acute phase protein C-reactive protein (CRP), while the cytokine TNF-α elicits metabolic effects within skeletal muscle, adipose tissue and bone.

Fig. 1 Generalised view of inflammation.
Characteristics of inflammatory conditions afflicting specific organ systems
Gut: celiac disease
Normal function of mucosal immunity
The gastrointestinal mucosa is an interface for communication between the individual and the external environment. The large mucosal surface (300–400 m2 in an adult human) is adapted with specialised structures for the chemical and immunological recognition of substances and organisms passing through the tract. Intestinal epithelial cells play a crucial role in detecting foreign substances and mediating host innate and adaptive mucosal immune responses. Activation of innate host defence mechanisms is based on the rapid recognition of conserved molecular patterns in microbes by preformed receptors, toll-like receptors, mainly expressed in the cell membrane and nucleotide-binding oligomerisation domain (NOD)-family (also known as caspase recruitment domain-family) receptors in the cytosol(Reference Aderem and Ulevitch1). In response to invading bacteria, the signals converge to transcription factors (NF-κB) and others), which initiate the transcription of genes responsible for the synthesis of proinflammatory proteins(Reference Elewaut, DiDonato and Kim2). Intestinal epithelial cells also express human leucocyte antigen (HLA) class II molecules, suggesting that they can function as antigen-presenting cells(Reference Maaser and Kagnoff3). On the other hand, non-pathogenic bacteria and innocuous food antigens may elicit other types of cytokine responses that are transmitted to underlying immunocompetent cells(Reference Haller, Bode and Hammes4). Normally, responses to non-pathogenic bacteria involve regulatory cytokines such as transforming growth factor (TGF)-β or IL-10, and are related with the induction of regulatory pathways of the immune system(Reference Borruel, Casellas and Antolin5, Reference Brandtzaeg, Farstad and Johansen6). Acquired immune responses develop in specialised lymphoid tissues. Gut-associated lymphoid tissue is located in three compartments: organised structures (Peyer's patches and lymphoid follicles); the lamina propria; the surface epithelium(Reference Shanahan, Johnson, Christensen, Jacobsen and Walsh7). The organised structures are the inductive sites for acquired immunity. These structures are covered by follicle-associated epithelium, which contains M-cells. These are specialised epithelial cells that transport micro-organisms and other antigens from the gut lumen into the organised lymphoid tissue. Antigens are presented to naïve T-cells by antigen-presenting cells such as dendritic cells (DC) and macrophages. In addition, luminal antigens may also be taken up and presented by epithelial cells. After priming, antigen-specific T-cell clones proliferate, but these T-cells may differentiate into Th1, Th2 or regulatory T-cells, with different effector capabilities(Reference Scott, Rognum and Midvedt8). Th1 or Th2 cells have a polarised cytokine secretion either of interferon (IFN)-γ, TNF-α and IL-2 (Th1), or IL-4, IL-5 and IL-13 (Th2). Such skewing of the adaptive immune response depends on the presence of micro-environmental factors, including ‘danger signals’ from microbial products, and eventually induces pro-inflammatory systemic-type immunity, with the potential for tissue damage. By contrast, regulatory T-cells produce IL-10 and TGF-β and lead to suppressive mechanisms that avoid local and peripheral hypersensitivity to innocuous antigens in food. Such mucosal tolerance to food antigens is an important adaptive immune function in view of the fact that as much as a ton of food may pass through the gut of an adult every year, resulting in substantial exposure to antigens. Thus, in the healthy state, the vulnerable gut mucosa exhibits virtually no proinflammatory response to food antigens(Reference Brandtzaeg9, Reference Cummings, Antoine and Azpiroz10) and contains very few hyperactivated T-cells.
Definition and description of celiac disease
Celiac disease is an immune-mediated disorder that affects primarily the small intestinal mucosa. The disease is triggered by the ingestion of gluten in genetically susceptible individuals. Strictly speaking, gluten is a protein component in wheat, but the term is collectively applied to disease-activating proteins in wheat, rye and barley. Celiac disease is characterised by chronic inflammation of the small intestinal mucosa that may result in atrophy of intestinal villi. The progressive destruction of the small intestinal mucosa causes malabsorption, and a variety of clinical manifestations, including diarrhoea, abdominal pain, vitamin and mineral deficiencies, iron-deficiency anaemia, osteoporosis, growth delay, skin lesions, neurological disorders, etc. Diagnosis of the disease requires examination of biopsies of small intestinal mucosa(Reference Mulder and Cellier11). The Marsh classification(Reference Marsh12) has been adopted to describe the progression of the abnormalities in the mucosa, from early stages with normal architecture and a lymphocytic infiltration of the villus epithelial layer up to total atrophy of the villi caused by chronic inflammation. A number of serologic tests are available commercially for identifying individuals who require an intestinal biopsy examination to diagnose celiac disease(Reference Rostom, Dube and Cranney13). The best markers are the detection in serum of anti-tissue transglutaminase IgA by ELISA, or anti-endomysial IgA by immuno-fluorescence. Both tests appear to have equivalent diagnostic accuracy as the tissue transglutaminase is the specific protein that is recognised by the IgA-endomysial antibody. Anti-gliadin antibody tests are no longer routinely recommended because of their lower sensitivity and specificity(Reference Rostom, Dube and Cranney13). The increasing use of serologic screening is leading to diagnosis in milder cases. It is presently recognised that, at certain points in time, the disease is not associated with obvious clinical signs and symptoms. Silent celiac disease is characterised by the presence of positive serologic markers (anti-tissue transglutaminase and/or endomysial antibody) and histological lesions on small bowel biopsy typical for celiac disease in an asymptomatic individual. In latent celiac disease, small bowel biopsy shows only minimal changes (increased intraepithelial lymphocyte infiltration) and anti-tissue transglutaminase or endomysial antibodies may be detected, but the characteristic feature is that the subjects develop symptoms and positive serologic and histological markers, while on a gluten-containing diet. Latent celiac disease precedes diagnosis of celiac disease or follows successful treatment of active disease with a gluten-free diet. Finally, active celiac disease is characterised by intestinal and/or extraintestinal symptoms, villus atrophy and strongly positive anti-tissue transglutaminase and endomysial antibodies.
Epidemiology of celiac disease
Population-based estimates of the incidence of confirmed celiac disease vary from two to thirteen new cases per 100 000 person-years. These rates have to be interpreted with caution because many patients diagnosed as adults are likely to have had several years of untreated celiac disease, and thus do not represent true new cases. The recent increase in the incidence rates has likely been due to increasing use of serologic screening leading to diagnosis in milder cases(Reference Rewers14). Although the prevalence of diagnosed celiac disease varies widely among different populations, the estimates of combined undiagnosed and diagnosed (latent, silent and active) celiac disease are remarkably similar, between 0·7 % and 2·0 %, in most European and non-European white populations, including those in Finland, Sweden, Germany, Italy, Spain, Israel, the United States, Argentina, Australia and New Zealand(Reference Rewers14, Reference Dube, Rostom and Sy15). However, celiac disease is virtually unknown in East Asian and African populations, whereas rates close to those in Europe have been reported from the Middle East and India(Reference Rewers14).
Genetic background appears to be a major risk factor for celiac disease. It is unequivocal that celiac disease is strongly associated with specific HLA class II genes that map to the DQ locus of the MHC used for cell-to-cell interaction during the process of antigen presentation(Reference Liu, Rewers and Eisenbarth16). The presence of specific alleles at the HLA-DQ locus appears to be necessary, although not sufficient, for the phenotypic expression of disease. Thus, virtually all affected individuals have either DQ2 (HLA-DQA1*05-DQB1*02) or DQ8 (HLADQA1*03-DQB1*0302) alleles, in comparison with the general population in which 39·5 % have either DQ2 or DQ8. However, only 3 % of individuals in the general population carrying DQ2 or DQ8 will develop evidence of celiac disease, suggesting that HLA typing could be used to identify genetic risk, but not for defining celiac disease(Reference Liu, Rewers and Eisenbarth16).
Environmental factors also play a major role. It is well established that celiac disease is strictly dependent on exposure to wheat gluten and related proteins in rye and barley. Thus, these cereal proteins are necessary causal factors both to initiate and to maintain the disease process. On the other hand, epidemiological studies have shown that breast-feeding is a protective factor. Introduction of gluten-containing foods while the infant is still breast-feeding reduces the risk for celiac disease(Reference Ivarsson17). The age of the infant at introduction of gluten-containing foods does not seem to be an independent risk factor, but it is well established that introduction of gluten-containing foods in large amounts, as compared with small or medium amounts, increases the risk for celiac disease. Infectious episodes affecting the gut mucosal barrier could potentially contribute to the development of celiac disease in genetically susceptible individuals(Reference Ivarsson17).
Description of the pathophysiological mechanisms involved in celiac disease
HLA class II molecules are expressed on the surface of antigen-presenting cells where they can bind and subsequently present ‘foreign’ peptides encountered in the extracellular environment to populations of naïve T-helper lymphocytes that recognise the DQ2- or DQ8–peptide complex. In the case of celiac disease, glutamine residues that remain in glutamine/proline-rich peptides following the intestinal digestion of ‘gluten’ are converted by tissue transglutaminase to negatively charged glutamic acid that binds to DQ2 or DQ8 molecules on antigen-presenting cells(Reference Kagnoff18). The DQ–‘gluten’ peptide complexes activate DQ2 or DQ8 restricted T-helper cells, respectively, that proliferate and produce mainly Th1-type cytokines, particularly IFN-γ. Of note, such gluten-activated Th1 cells were isolated from the small intestinal mucosa of patients with celiac disease, but they are not found in the intestinal mucosa of individuals without celiac disease, who also have the relevant disease-associated DQ2 or DQ8 HLA class II molecules(Reference Kagnoff18). Secretion of Th1 cytokines such as IFN-γ and TNF-α activates the release of enzymes such as matrix metalloproteinases that can damage the intestinal mucosa, with a loss of villus structure(Reference Kagnoff18). These cytokines increase epithelial permeability and proinflammatory cytokine production, which in turn will increase the passage of gluten peptides and peptide binding to DQ2 and DQ8 molecules on antigen-presenting cells, leading to a chronic feedback of the inflammatory process as long as gluten peptides are present in the intestinal lumen.
A strict gluten-free diet for life is the only accepted treatment for celiac disease(Reference Sollid and Khosla19). Successful outcomes are highly dependent on compliance to such a diet. Advanced celiac disease may not respond to a gluten-free diet, and these refractory cases are being treated with immunosuppressant drugs, including azathioprine and anti-TNF therapies with variable results (Fig. 2).
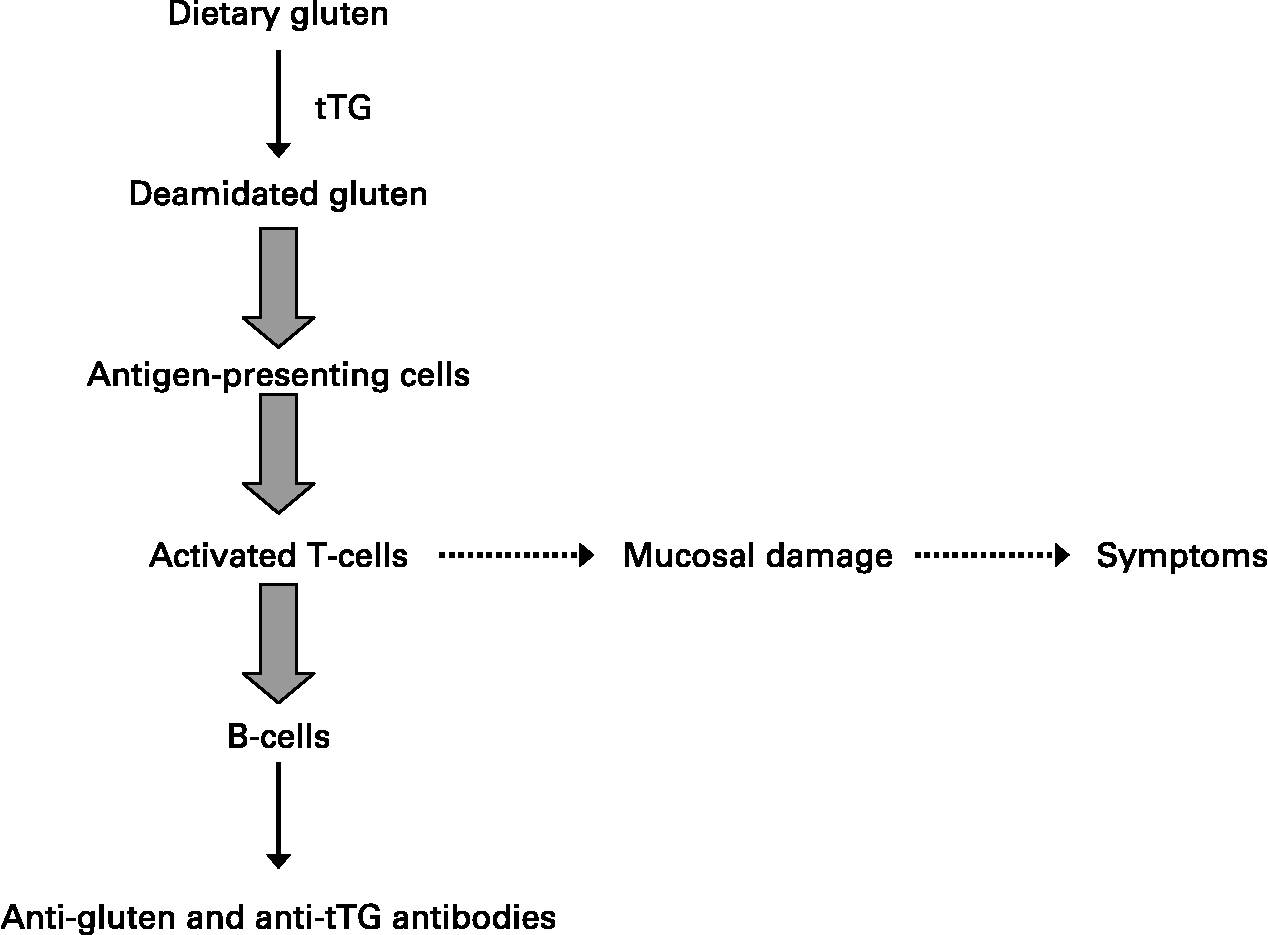
Fig. 2 Generalised scheme for the development of celiac disease.
Gut: inflammatory bowel diseases
Definition and description of inflammatory bowel diseases
Ulcerative colitis (UC) and Crohn's disease (CD) are the two main forms of inflammatory bowel disease (IBD). UC and CD are both acute and chronic disabling inflammatory illnesses, whose aetiologies are unknown. Present opinion holds that IBD are multifactorial conditions, in which they involve both genetic and environmental components, with the final outcome being determined by an aberrant immune response to normal commensal microbiota in individuals who have a weakened epithelial barrier(Reference Duchmann, Kaiser and Hermann20–Reference Shanahan22). IBD is incurable, but typical maintenance treatments involve the use of immunosuppressant and anti-inflammatory drugs, antibiotics and surgery. An important factor differentiating CD from UC is that it can affect any part of the gastrointestinal tract, not just the large bowel as in UC. Whereas CD is characterised by patchy, transmural inflammation with inflammatory processes occurring deep in the tissues, the inflammatory response in UC is primarily located in the colonic mucosa and submucosa resulting in severe tissue damage, ulceration and haemorrhage(Reference Carter, Lobo and Travis23). In both forms of IBD, large infiltrates of neutrophils can be seen in the inflamed tissues, together with the presence of microabscesses in the lamina propria. The main clinical feature of UC is bloody diarrhoea; other symptoms such as colicky abdominal pain and an urgency to defaecate may also be present, whereas in CD the symptoms can be more heterogenous and include diarrhoea, abdominal pain, weight loss and malaise(Reference Carter, Lobo and Travis23). Both CD and UC are associated with a 5-fold increased risk of developing colon cancer.
Epidemiology of inflammatory bowel diseases
Up to two million people are affected by IBD globally, with the disease being mainly associated with industrialised countries in the northern hemisphere. The incidence of CD is increasing in the UK and is about five to ten per 100 000 per annum, with a prevalence of 50–100 per 100 000, although this is believed to be an underestimate(Reference Carter, Lobo and Travis23). Overall, the incidence and prevalence of UC is approximately twice that of CD. IBD usually starts early in life with the greatest onset being in young adults aged between 15 and 25 years, but it can affect people of any age, with 15 % of people aged over 60 years at diagnosis.
Although a genetic component is known to be involved in IBD, with both CD and UC sharing some susceptibility genes, there is stronger evidence for a genetic link in CD(Reference Farrell and Peppercorn21, Reference Shanahan22). In CD, a 44 % concordance exists between monozygotic twins, and a 4 % concordance between dizygotic twins. Between 4·5 and 16·6 % of CD patients have the disease already in the family, suggesting a polygenetic inheritance. A mutation in the NOD2/caspase recruitment domain-15 (called IBD-1) gene on chromosome 16 has been found in 30 % of patients with CD(Reference Ogura, Bonen and Inohara24). NOD2 is a cytoplasmic receptor for muramyl peptide found in bacterial cell walls, which may result in CD patients with this mutation having a reduced ability to clear invasive bacteria. A higher incidence of UC has been found in Jewish than in non-Jewish families, and Ashkenazi Jews born in the West have been found to have a higher incidence of CD than those born in Israel or non-Ashkenazi Jews. Largely uncategorised environmental factors are thought to predispose towards IBD. However, the change to a more Western-style diet has been suggested as a link in IBD development. Smokers are more likely to develop CD and have more aggressive disease, with more severe relapses than non-smoking CD patients, although smoking has been shown to be protective in UC. Physiological stress, particularly long-term stress, may also be a precipitating factor in IBD. Stress can also bring about relapses in animal models of colitis and cotton-top tamarinds spontaneously develop colitis when kept in long-term captivity. Childhood illnesses and not being breast-fed are also thought to incur increased risk of developing IBD; however, appendectomy in early life can prevent the development of UC.
Description of the pathophysiological mechanisms involved in inflammatory bowel diseases
There is considerable evidence for microbial involvement in IBD. For example, while germ-free animals do not manifest an inflammatory response, knock-out or transgenic mice with genetic susceptibilities to IBD only acquire characteristic lesions when repopulated with normal commensal bacteria(Reference Taurog, Richardson and Croft25). Human studies have shown that UC patients have increased levels of mucosal IgG against the normal colonic microflora, as well as increased antibody production against strictly anaerobic bacteria. UC also starts in the area of the gut with the highest concentration of micro-organisms. A number of different bacteria have been implicated in IBD in human subjects and animals, in various studies, but none have been definitively shown to be aetiologic agents in human subjects. The role of mucosal bacteria may be important due to their ability to interact more directly with the host immune system(Reference Macfarlane, Furrie and Kennedy26, Reference Furrie, Macfarlane and Kennedy27). Nevertheless, CD patients have been shown to have higher numbers of bacteroides and lower numbers of bifidobacteria in faecal(Reference Seksik, Rigottier-Gois and Gramet28) and mucosal samples, while higher numbers of peptostreptococci and lower numbers of bifidobacteria have been found on the mucosa in patients with UC(Reference Macfarlane, Furrie and Cummings29). Low numbers of bifidobacteria may be of significance in IBD because some species exhibit strong immunomodulatory properties(Reference Famularo, Moretti, Marcellini and Fuller30). This dysbiosis in mucosal bacterial populations in IBD, with the loss of beneficial commensal species, and perhaps a switch to a more pro-inflammatory phenotype, may explain why probiotic bacteria have been shown to be useful, in some cases, in the treatment of some forms of IBD, such as UC and pouchitis(Reference Furrie, Macfarlane and Kennedy27, Reference Macfarlane, Furrie and Cummings29, Reference Gionchetti, Rizzello and Venturi31).
In IBD, the interaction between the mucosal immune system and the commensal microflora in the gut is disturbed and dysregulation of the immune system occurs. The T-cell response profiles associated with UC and CD are different in that a Th1 pattern of cytokine formation develops in CD with increased production of TNF-α, IFN-γ, IL-12, IL-6 and IL-1β, whereas UC more resembles a modified Th2 profile, where cytokines including IL-5 and IL-10 are upregulated, although IL-4 is not(Reference Farrell and Peppercorn21). In addition to this change in cytokine profile, intestinal B lymphocytes produce large amounts of IgG. TNF-α is expressed in the intestinal mucosa of patients with IBD and triggers inflammation via an NF-κB-dependent signalling cascade. Most of the pro-inflammatory responses in IBD are transcriptionally regulated by NF-κB and many of the cytokines involved act on signal transducers and activators of transcription (STAT) family(Reference Musso, Dentelli and Carlino32). IL-12 can act on CD4+T-cells to induce differentiation into Th1 effector cells and activate the signalling proteins STAT-3 and STAT-4, which in turn leads to the production of IFN-γ. Animal studies have shown that STAT-4-deficient mice have impaired IFN-γ production, and do not develop colitis, whereas mice overexpressing STAT-4 have diffuse infiltration of IFN-γ and develop gut inflammation. STAT-3 signalling has been found in UC and CD where it has been shown to be confined to areas of active inflammation, infiltrating macrophages and T-cells. STAT-3 induces transcription of the pro-inflammatory cytokine IL-6, which can increase resistance of T-cells to apoptosis lengthening the chronicity of CD, due to the accumulation of active T-cells. Increased numbers of blood mononuclear cells have also been found in IBD, which may lead to extraintestinal manifestations, while the production of free radicals from macrophages contributes to cellular damage. Other factors implicated in CD include generation of matrix metalloproteinases, such as collagenases and stromelysins, which can degrade extracellular matrices, cause ulceration and result in tissue destruction(Reference Shanahan22, Reference von Lampe, Barthel and Coupland33). High levels of extracellular matrix metalloproteases, which can be upregulated by pro-inflammatory cytokines, have been shown in areas of tissue injury and foci of ulceration in CD patients. Group II phospholipase in serum and colonic mucosa has also been shown to be increased in patients with CD, which can be inhibited by steroids and anti-inflammatory drugs. Corticosteroids are the main therapy used for treatment of active IBD, to downregulate the immune response, and allow the mucosa to heal. More recently, therapies involving the use of monoclonal antibodies such as Inflixamab against TNF-α have been found to be effective in inducing remission in CD.
Lungs: airways and the allergic inflammation
Normal functioning of the airways
The airways bring air into the alveoli of the lungs, where air and blood are separated only by the alveolar membrane that is about 0·001 mm thick. The turbulence of the air passing through the upper airways is such that most particles larger than 2 μm in diameter are caught on the mucosal surface in the nasal cavity. This is beneficial with regard to starting an immune response against foreign material, because the back of the nasal cavity and the upper pharynx are rich in immune cells and lymphoid tissue. The trachea splits into two main bronchi that continue to branch out into bronchi and bronchioli, to end in the so-called alveolar sacs and alveoli. Alveoli have a good blood supply and are extremely thin-walled to allow gas exchange between blood and air. From the alveoli, the anatomical structure of the airways gradually changes towards the bronchi. The basic components are the epithelium that covers the inside of the airway tubes, and under the epithelium, the submucosa and then a layer with smooth muscle that can contract and thereby narrow the airways. In certain areas, the epithelial surface against the airway lumen has fine hair-like structures called cilia, which have a contractile apparatus and a synchronised beat so that mucus and particles will be moved up towards the larger airways and the larynx (‘the mucociliary escalator’).
Normal function of inflammatory responses to maintain homeostasis in the airways
The alveolar membrane (140 m2 surface area) is protected by three major, integrated defence systems along the airways. Firstly, the anatomy of the airways provides ‘mechanical’ defence, conditioning and cleaning the air as it passes through the nose. The mucociliary escalator helps remove particulate material. Secondly, the inflammatory response mediated by several cell types results in a rapid capacity to eliminate intruding microbes as well as foreign material and debris, but may become long-lasting if the material is not properly removed (in this case lung disease may result). Finally, the specific immune response is highly effective at eliminating microbes and other foreign material. The human lung is exposed daily to between 10 000 and 20 000 litres of ambient air containing large numbers of particles and gases that may cause oxidative stress and inflammation. Not surprisingly, the lungs have several systems to counter oxidative stress(Reference Rahman, Biswas and Kode34).
Description of inflammatory airway and lung diseases: rhinoconjunctivitis, asthma and chronic obstructive pulmonary disease
The main diseases of the airways to be addressed are allergic rhinoconjunctivitis, allergic asthma and chronic obstructive pulmonary disease (COPD). These three are usually described as separate entities, but it is important to realise that there is a strong link between allergic rhinoconjunctivitis and asthma, and that asthma and COPD may be part of a continous spectrum of disease. At the one end of this spectrum would be allergic asthma with near-complete reversibility of bronchial obstruction and little inflammatory change and at the other end COPD with nearly no reversibility of airflow limitation, and pronounced inflammation and destruction of lung tissue. If asthma and COPD are considered as separate diseases, then many patients present components of both diseases to varying degrees. These components of airway disease include signs like variable airflow limitation, airway hyper-responsiveness, chronic airflow limitation and airway inflammation with different characteristics, as well as bronchiectasis and emphysema. Symptoms include chest tightness, wheezing, dyspnoea, cough and sputum and are all non-specific, since they can result from several disease components. Measures of lung function and inflammation are necessary to be certain as to which condition the symptoms are associated with(Reference Hargreave and Parameswaran35). Thus, many patients with COPD and asthma can be readily distinguished from one another, but many patients also have features of both disorders(Reference Elias36).
Allergic rhinoconjunctivitis is a fairly ‘pure’ allergic disease, in which the classical anti-allergen IgE-mediated release of histamine and other mediators from mast cells triggers the classical symptoms and signs of itching, sneezing, oedema, influx of eosinophils and watery mucous production. In contrast to allergic asthma, in rhinitis, there is no smooth muscle constriction narrowing the airways (narrowing is caused by swelling of the mucosa), and no significant tissue destruction. Subjects with allergic rhinoconjunctivitis are often found to have bronchial hyper-reactivity, and may later develop asthma.
Asthma is considered a chronic inflammatory disease of the lungs. Asthma is traditionally classified into allergic and non-allergic asthma. Allergic asthma is considered to be the most common form of asthma in children, while in adults asthma without known allergen triggers is more common. However, the distinction depends on the demonstration of triggering allergens, and is, therefore, somewhat unclear. It may be argued that asthma and allergy at least sometimes may be parallel but separate conditions (e.g. ‘non-allergic’ asthma in an individual that also has allergies). With both types of asthma, various ‘non-specific’ irritants may aggravate asthma and trigger an asthmatic attack, and the allergen-triggered allergic inflammation can be looked upon as one of several such asthmagenic stimuli. Asthma has chest tightness, wheeze, cough and dyspnoea as prominent symptoms, and is functionally characterised as reversible bronchial obstruction, caused by contraction of the smooth muscle layer in the mucosa of the bronchi, by mucous production, mucosal oedema and mucosal inflammation. Airway hyper-responsiveness (oversensitivity and overreactivity to stimuli) is typically present in asthma, but is not an obligatory feature. Similarly, bronchial hyper-responsiveness is often found in individuals without manifest asthma. A prominent cell in the asthmatic inflammation is the eosinophil, together with lymphocytes. Granulocytes other than eosinophils may be present to varying degrees. The inflammation may lead to destruction and shedding of the epithelial cell layer. Over time, structural changes take place in asthma, so-called remodelling, inflammation will become permanent and more severe, and reversibility of the airways obstruction will become less complete. Chronic severe asthma may thus show features of COPD.
The definition of COPD rests much on functional criteria, with non-reversible or incompletely reversible airflow obstruction accompanied by shortness of breath, cough and sputum production, and intermittent exacerbations. However, some reversibility of airways obstruction may be present, and bronchodilators are the basic medication used in COPD. Underlying the symptoms are chronic lung inflammation and progressive, often pronounced tissue destruction. In addition to pulmonary manifestations, other organ systems may be affected in COPD. Systemic manifestations of COPD include weight loss, nutritional abnormalities and musculoskeletal dysfunction(Reference Agusti, Noguera and Sauleda37–Reference Gan, Man and Senthilselvan39).
Epidemiology of allergic rhinoconjunctivitis, asthma and chronic obstructive pulmonary disease
Allergic rhinitis is triggered by allergens, but symptoms may be increased by air pollution. Allergic rhinitis may be constant (perennial) if the eliciting allergen is constantly present (e.g. mite allergens), seasonal if the allergen shows seasonal variation (e.g. in pollen allergy in the Nordic countries) or episodic (e.g. a person allergic to cats with low habitual exposure, who visits a home with cats). Allergic rhinitis starts in infancy and peaks in childhood and adolescence. Atopic individuals constitute the group at risk. In 80 % of cases, the symptoms develop before the age of 20 years. Severity varies from trivial symptoms to periodically incapacitating disease. Allergic rhinitis may be associated with other upper-airway inflammatory conditions(Reference Phipatanakul40, Reference Scadding, Mitchell, Albert, Spiro and Jett41). A large international study showed considerable variation in prevalence between countries, from 0·8 % to 14·9 % in 6- to 7-year olds, and from 1·4 to 39·7 % in 13- to 14-year olds(Reference Strachan, Sibbald and Weiland42). There has been a general pattern of increased prevalence in the industrialised compared with the developing countries. Cross-sectional studies with similar methodology suggest a rising prevalence(Reference Maziak, Behrens and Brasky43).
Asthma severity varies from barely noticeable symptoms to lethal exacerbations. Prevalence of asthma can vary 3-fold in the same population depending on the diagnostic criteria used, and prevalence data should be interpreted cautiously(Reference Postma, Kerstjens, Ten Hacken, Albert, Spiro and Jett44). Atopic individuals constitute the major risk group. However, as already mentioned, asthma is also frequently diagnosed in non-allergic individuals. Asthma is not only common in children but also develops in adult life. Diagnosis in small children is uncertain. For all age groups, incidence varies between countries from 2·7 to 4 per 1000 individuals per year. After 25 years of age, it has been estimated at 2·1 per 1000 individuals(Reference Postma, Kerstjens, Ten Hacken, Albert, Spiro and Jett44). Before puberty, asthma is more common in boys than girls, but after puberty this sex difference is reversed. Prevalence has been extensively studied in the last 30 years, and one major study is the International Study of Asthma and Allergies in Childhood(45, 46). The present study of nearly half a million children aged 13–14 years in 56 countries showed large variability between countries. The highest 12-month prevalence was found in the UK, Republic of Ireland, Australia and New Zealand (up to 36·8 %), and the lowest in Eastern European countries, Indonesia, China, India and Ethiopia (the lowest 1·6 %). A number of studies have found an increasing incidence and prevalence over the last decades(Reference Postma, Kerstjens, Ten Hacken, Albert, Spiro and Jett44, Reference Russel47). However, some fairly recent studies have indicated that in some regions(Reference Zollner, Weiland and Piechotowski48), but not others(Reference Lodrup Carlsen, Haland and Devulapalli49), the increase may have levelled out.
In 1990, COPD was the twelfth leading cause of morbidity and the sixth leading cause of death worldwide. Of all the major diseases, COPD is the one whose burden is rising the fastest, and it is projected to become the fifth leading cause of disability and the third leading cause of death by 2020(Reference Murray and Lopez50). COPD is strongly associated with cigarette smoking, but also occurs in non-smokers, and individuals vary greatly in their susceptibility to the effect of tobacco smoke. COPD is markedly underdiagnosed and frequently undermanaged, perhaps because it has been considered irreversible and that treatment has little to offer. However, COPD may be partially reversible and clinical responses to treatment do occur(Reference MacNee, Albert, Spiro and Jett51).
Description of the pathophysiological mechanisms of lung disease, with an emphasis on inflammation
Environmental triggers
Asthma appears to be caused by environmental factors, such as allergens, irritants and infections, in genetically predisposed individuals. Once asthma has become manifest, essentially the same agents may trigger worsening of the disease and precipitate serious asthmatic attacks, as well as contribute to the development of chronic disease. A host of triggering as well as some protective factors are known, but the basic understanding of why and how asthma develops is still rudimentary. A major factor contributing to the development of allergy and asthma is the so-called Western lifestyle. This is illustrated by the 2- to 3-fold higher prevalence of asthma in former West Germany compared with East Germany, and Hong Kong compared with nearby cities in China(Reference Leung and Ho52, Reference von Mutius, Martinez and Fritzsch53). Gradients in the prevalence of allergic disease corresponding to social gradients are also found within individual countries, and allergic diseases have been described as a price paid for wealth and a high standard of living. Environmental and lifestyle factors have changed in a broad sense over the last decades in industrialised countries, but it is not known which specific conditions in the more affluent, industrialised or urbanised lifestyle drive the development of asthma and allergy. Also, asthma is a major health problem among the poor populations living in deprived inner city districts and the homeless in USA, with a very high prevalence of severe asthma, and prevalence is also high in some other underprivileged regions e.g. in South America. Asthma therefore does not, in all places, fit the ‘hygiene hypothesis’ model of more allergic disease among the ‘clean and rich’ and less among the poor (see later).
Allergen exposure will trigger asthma once the disease has become manifest, as will a wide array of non-specific irritants in the occupational setting, indoor environmental factors (e.g. second-hand tobacco smoke, strong odours and perfume) and outdoor air pollution factors like fine particulate matter, ozone and oxides of nitrogen. Also, physical exercise and psychological stimuli may precipitate asthma(Reference Postma, Kerstjens, Ten Hacken, Albert, Spiro and Jett44).
For the causation of asthma, loss of protective environmental factors may be as important as asthma-provoking factors. The observation that the risk of asthma is reduced with increasing number of older siblings was explained by the Th1–Th2 pardigm of immune responses (see later) and the so-called hygiene hypothesis. In its original form, this hypothesis stated that younger siblings got more infections and got them earlier than the older siblings, and that the infections stimulated the development of Th1 immunity and counteracted the development of allergy-associated Th2 responses(Reference Strachan54). Although the focus has moved away from the hygiene hypothesis in the original form(Reference Umetsu55–Reference Cullinan57), it is still considered that ‘something with micro-organisms’ is protective in relation to asthma and allergy(Reference von Mutius58). Observations that more or less indirectly support this include the inverse relationship between family size and asthma(Reference von Mutius, Martinez and Fritzsch59), the protective effect of early placement in day care settings(Reference Ball, Castro-Rodriguez and Griffith60), the protective effect of exposure to farm animals(Reference Riedler, Braun-Fahrlander and Eder61), the protective effect of endotoxin exposure (allergy more than asthma)(Reference von Mutius58), the reduced prevalence of asthma among anthroposophic Steiner school attendants(Reference Floistrup, Swartz and Bergstrom62), the protective effect of extensive exposure to cats and dogs(Reference Platts-Mills, Vaughan and Squillace63), the association between intestinal flora and atopic manifestations(Reference Bjorksten, Naaber and Sepp64) and the association between early use of antibiotics and increased incidence of allergy(Reference McKeever, Lewis and Smith65). Other hypotheses of asthma causation include reduced intake of antioxidants from foods like fruit and vegetables(Reference Wong, Ko and Hui66, Reference Chen, Hu and Seaton67) and an altered balance of fatty acids in the diet(Reference Mickleborough and Rundell68). Finally, obesity(Reference Beuther and Sutherland69) and physical inactivity(Reference Shaaban, Leynaert and Soussan70) are associated with asthma and bronchial hyper-reactivity. Whatever the explanation, asthma must be said to represent ‘an epidemic of dysregulated immunity’(Reference Umetsu, McIntire and Akbari71).
For COPD, the major known environmental trigger is tobacco smoke. Also occupational chemicals and particulate matter may contribute to the development of the disease, as well as indoor and outdoor air pollution. Diet has been reported to be a modifying factor, with fruit and vegetable consumption having a protective effect(Reference Watson, Margetts and Howarth72).
Perpetuating factors
In allergic diseases, like allergic rhinoconjunctivitis and allergic asthma, repeated and chronic allergen exposure will keep the disease active and worsening. Typically, general hypersensitivity and hyper-reactivity also to non-specific stimuli develop. These will contribute to perpetuating the disease, and viral infections and environmental pollutants (e.g. tobacco smoke and diesel exhaust particulates) will worsen the disease and may precipitate acute attacks of severe asthma. Largely, the same non-specific stimuli as seen in asthma, but in particular tobacco smoke, will contribute to perpetuating and accelerating COPD.
Mechanisms and mediators
Alveolar macrophages in the lumen of the airways are first-line defence cells, capable not only of engulfing foreign material to destroy it, but also of secreting several signal molecules (cytokines) that serve to attract and activate other cells to create inflammation. Macrophages can remove residual material after phagocytosis in two ways. They can go through the bronchiolar and alveolar walls to enter the lymphatic system carrying fluid and cells to the numerous lymph nodes located along the trachea and bronchi, or move out along the airways helped by the mucociliary escalator mentioned earlier, to be swallowed into the stomach or spat out. Particulate matter that is not dissolved may remain in the lymph nodes for a long time.
Epithelial cells are themselves active participants in immunoregulation and inflammation by secreting a number of inflammatory and immunoregulatory molecules. Furthermore, specialised cells (goblet cells) secrete protective mucus to cover the epithelial surface, and so-called type 2 cells secrete a family of molecules collectively referred to as surfactant. The surfactant has a number of beneficial functions, among others to regulate immune responses and inflammation(Reference Kingma and Whitsett73).
In the submucosa, various cells of the specific and non-specific immune systems can be found; among them, mast cells are important also in allergic reactions. The blood vessels bring various immune cells to the lung submucosa and epithelium. These include various subgroups of T and B lymphocytes and natural killer cells, monocytes, neutrophils, eosinophils and other cells. These various cells, in addition to performing defence tasks by cell-to-cell contact, carry with them or produce on-site two groups of molecules, signal molecules and defence molecules (with somewhat overlapping functions). Signal molecules more or less specifically regulate functions in other cells. For example, CD4+T lymphocytes can secrete the cytokines IL-13, IL-4 and IL-9 to stimulate mucous production by epithelial cells, IL-13 and IL-9 to stimulate bronchial muscle contraction, IL-4 and IL-13 to stimulate antibody production by B lymphocytes, IL-3, IL-4, IL-5, IL-9, IL-13 and granulocyte–macrophage colony stimulating factor to attract and activate various other cells (sometimes in a very specific way), and IL-3, IL-4, IL-9 and IL-13 to stimulate the development of other cells. Accumulation and activation of cells is an important feature of the inflammatory response, and the cytokines mentioned will more or less contribute to such a response. Some other cytokines are particularly important for inflammation, like TNF-α, IL-6, and IL-8 and IL-17, while IL-10 tends to downregulate inflammation as well as immune responses – an important function to protect against self-destruction. Other defence molecules are designed to directly destroy the intruders: lysozyme; lactoferrin; complement; α-1 antitrypsin; cationic proteins; lysophospholipases; catalases; non-specific esterases; others. The various granulocytes and the mast cells have in their cytoplasm granules containing preformed mediators that are released in the ‘combat’ situation.
At the heart of the allergic reaction is the interaction between IgE molecules bound to specific receptors on the membrane of mast cells and their corresponding allergens. When the IgE molecules are cross-linked by allergen, the mast cell is triggered to release the potent inflammatory mediators contained in its cytoplasmic granules, and the allergic inflammatory response develops. This response has two phases, an early virtually immediate reaction and a late response developing after some 6 to 8 h. Mast cells are the key cells in the early response, while eosinophils are the predominant cell in the late response. Eosinophils are thought to play a central role in allergic rhinitis as well as asthma(Reference Scadding, Mitchell, Albert, Spiro and Jett41, Reference Van Wetering, Hiemstra, Rabe, Albert, Spiro and Jett74). Asthma historically changed from being looked upon as a disease of the smooth musculature of the bronchi to being a Th2-dominated chronic inflammatory disease, with the eosinophil as the typical infiltrating cell. Other cells that regulate Th2 responses, asthma and allergy are NKT cells(Reference Akbari, Stock and Meyer75), and the various types of regulatory T-cells (Treg cells) that have the capacity to inhibit the effector function of Th2 and Th1 cells(Reference Erin, Leaker and Nicholson76). Increased levels of the Th2 cytokines IL-4, IL-5, IL-9 and IL-13 have been demonstrated in the asthmatic airway(Reference Ray and Cohn77). The Th2-driven inflammation has two arms, one via B-cells activated by IL-4 to produce IgE, which triggers the mast cell-mediated allergic inflammation, and the other via IL-4-, but mainly via IL-13-mediated direct effects on epithelium and bronchial smooth muscle(Reference Barrios, Kheradmand and Batts78). However, whereas the Th1/Th2 concept revolutionised immunological thinking when first proposed in 1987, it has been challenged over the past several years for several reasons(Reference Umetsu56, Reference Steinman79). Both Th1 and Th2 cells are pro-inflammatory, and Th1 cells do not always downregulate Th2 cells, but may instead exacerbate Th2-mediated diseases(Reference Hansen, Berry and DeKruyff80–Reference Heaton, Rowe and Turner82). It is of interest that the Th1 cytokine IFN-γ, now considered an aggravating factor in severe asthma, is also considered to be important in COPD(Reference Saetta, Di and Turato83). Recently, TNF-α has also been reported to play an important role in severe asthma(Reference van Oosterhout and Bloksma84). That Th1 cells play a role in asthma development is also illustrated by the finding that the Th1 transcription factor T-bet controls features of asthma. In the absence of T-bet (e.g. in T-bet-deficient mice), airway remodelling and airway hyper-reactivity develop spontaneously(Reference Finotto, Neurath and Glickman85). Additionally, it has been demonstrated that different asthmatic individuals display different ‘individualised’ cytokine patterns, caused probably both by different genetic factors and by differences in environmental exposure(Reference Heaton, Rowe and Turner82). Neural mechanisms are central in the asthmatic inflammation(Reference Nassenstein, Schulte-Herbruggen and Renz86, Reference De Swert and Joos87), but may also have a capacity to downregulate inflammation as has been shown in gastrointestinal inflammation(Reference Tracey88).
A number of genes have been implicated in asthma, e.g. ADAM33(Reference Van Eerdewegh, Little and Dupuis89). It is estimated that more than a dozen polymorphic genes regulate features of asthma like the inflammatory response, IgE synthesis, cytokine and chemokine production, airway remodelling and airway function(Reference Fahy, Corry and Boushey90).
On the molecular level, oxidative stress appears to play an important role in asthma(Reference Lee, Park and Park91, Reference Fujisawa92), and the endogenous antioxidant capacity of the lungs has been found to be defective in asthmatic patients(Reference Dworski93). Oxidative stress is caused by ROS. The inflammatory cells recruited into the asthmatic airways have a capacity to produce ROS as part of normal antimicrobial defence. Also, exogenous factors precipitating asthma, not only ozone and oxides of nitrogen, but also fine particulate matter, appear to cause oxidative stress(Reference Chan, Wang and Li94, Reference Vinzents, Moller and Sorensen95). In animal models, factors that inhibit increased ROS generation have been found to reduce inflammation and hyper-reactivity, two important components of asthma(Reference Lee, Link and Baluk96).
While irreversible airflow limitation is a functional hallmark of COPD, the lesion is histopathologically characterised by inflammation and tissue destruction. Dominating inflammatory cells are neutrophils, macrophages and CD8+-T lymphocytes and IFN-γ is an important cytokine(Reference Saetta, Di and Turato83). Oxidative stress appears to be important in the inflammation and tissue damage in COPD. Environmental agents linked to COPD, like tobacco smoke and particulate matter, induce oxidative stress and inflammation. Increased levels of markers of oxidative stress have been found in exhaled breath condensates of COPD patients(Reference Kharitonov and Barnes97–Reference Paredi, Kharitonov and Barnes99), and in a mouse model of tobacco smoke-induced COPD tissue, damage due to oxidative stress was observed. Corticosteroid resistance in COPD has also been related to oxidative stress. An imbalance between protease and anti-protease enzymes is thought to be of importance for the development of the emphysema component of COPD(Reference Shapiro100), and this is supported by findings in experimental models(Reference D'Armiento, Dalal and Okada101).
Joints: rheumatoid arthritis
Normal function of inflammatory processes to maintain homeostasis
Controlled inflammation as a result of various triggers is likely to occur in normal joints. Possible triggers are local mechanical stress and transient infection. Local mechanical stress leads to induction of cellular stress proteins, which can be targeted by stress protein-specific regulatory T-cells(Reference van Eden, van der Zee and Prakken102). Besides genetic factors and infection, there is some conflicting data on failing T-cell regulation as a critical factor in the aetiology of rheumatoid arthritis (RA)(Reference van Amelsfort, Jacobs and Bijlsma103).
Definition and description of rheumatoid arthritis
RA is a common autoimmune disease characterised by chronic inflammation of the synovium of diarthrodial joints(Reference Lee and Weinblatt104). It can lead to long-term joint damage, resulting in chronic pain, loss of function and disability. Primarily, the small joints of the extremities are affected, but as the disease progresses joints become involved. The chronic inflammatory process changes the cellular composition (cellular infiltration) and the gene expression profile of the synovial membrane, resulting in hyperplasia of the synovial membrane, which causes structural damage in cartilage, bone and ligaments. Extra-articular disease affecting a variety of organs is a significant factor in morbidity and mortality of RA(Reference Pincus and Callahan105). The severity of RA encompasses a wide spectrum, ranging from self-limiting disease to chronic progressive disease, causing varying degrees of joint destruction and extra-articular organ involvement.
Epidemiology of rheumatoid arthritis
RA occurs in 0·5–1·0 % of the population worldwide(Reference Silman and Pearson106). RA is about two to three times more common in women than men. No increased incidence of RA has been noted over recent decades. In fact, decreases were documented, although this might have been artificially caused by a changed clinical classification of diseases previously characterised as RA(Reference Uhlig and Kvien107). Juvenile idiopathic arthritis (JIA) is one of the most common autoimmune diseases in childhood(Reference Woo and Wedderburn108). Although JIA has strong histopathological similarities to RA, it forms a separate clinical entity. Whereas untreated RA generally is progressive, JIA consists of different subtypes with striking differences in both severity and outcome. One subtype is persistent oligoarticular JIA, characterised by a remitting and often spontaneously self-limiting course. As a result of this, persistent oligoarticular JIA is unique among all autoimmune diseases. The self-limitation of an autoimmune process is often seen in experimental animal models but hardly ever in human subjects and seems to suggest the active involvement of immunoregulatory processes, such as regulatory T-cells. Indeed, the frequency of CD4+CD25+ regulatory T-cells in the peripheral blood and in the synovial fluid of JIA patients has been found to correlate with the clinical course of the disease(Reference de Kleer, Wedderburn and Taams109, Reference de Kleer, Kamphuis and Rijkers110). Neither hygienic nor unhygienic living conditions are associated with the risk of developing juvenile RA(Reference Nielsen, Dorup and Herlin111). Although the causes of RA remain elusive, the general consensus is that factors contributing to its occurrence and course (clinical heterogeneity) are probably both genetic and environmental. The main risk factors for the disease include genetic susceptibility, sex and age, smoking and infectious agents. In addition, hormonal, dietary, socio-economic and ethnic factors seem to contribute(Reference van der Helm-van Mil, Wesoly and Huizinga112). For RA, the main predisposing genetic factor is HLA-DR4. HLA-DR4 predisposes for a more severe progressive disease course. Besides HLA-DR4, other genetic factors have recently been discovered in RA, such as the genetic polymorphisms in the lymphoid protein tyrosine phosphatase (LYP), associated with both type 1 diabetes and RA(Reference Begovich, Carlton and Honigberg113). As LYP is responsible for the tuning of T-cell activation, genetic variation of LYP will lead to variation in overall lymphocyte reactivity. The presence of anti-cyclic citrullinated peptides (CCP) antibodies (see later) seems to be an indicator for a higher grade of joint destruction and disease persistence. Smoking was found to be a risk factor for the development of anti-CCP antibodies. Anti-CCP positive RA may be a distinct disease entity that can react differently to treatment(Reference van der Helm-van Mil, Verpoort and Breedveld114).
Pathophysiological mechanisms of rheumatoid arthritis
RA involves many elements of the immune response. The synovium (or synovial membrane) is normally a relatively acellular structure consisting of one or two layers of synoviocytes. In RA, the synovium becomes hypertrophic and oedematous. Angioneogenesis, recruitment of inflammatory cells due to production of chemokines, local retention and cell proliferation contribute to the accumulation of cells in the inflamed synovium. Locally expressed degradative enzymes digest extracellular matrix and destroy articular structures(Reference Firestein115). The synovial membrane that extends to the cartilage and bone is known as pannus. It actively invades and destroys the periarticular bone and cartilage at the margin between synovium and bone. T-cells are actively involved in the pathogenesis of RA. Activated T-cells that are abundantly present in the inflamed joints of RA patients can stimulate other cells (e.g. B-cells, macrophages and fibroblast-like synoviocytes)(Reference Panayi, Lanchbury and Kingsley116). These T-cells are found to participate in the complex network of cell- and mediator-driven events leading to inflammation and joint destruction. B-cells play several critical roles in the pathogenesis of RA. They are the source of autoantibodies being produced in RA and contribute to immune complex formation (rheumatoid factors reactive with the constant region of their autologous IgG molecules) and complement activation in the joints(Reference Weyand, Seyler and Goronzy117). Multiple other autoantibodies have been found in RA, with recent interest focused on those directed at CCP(Reference Bridges118). Antibodies that are directed to citrullinated protein(Reference Vossenaar and van Venrooij119) detect CCP in many different proteins and are present in about 80 % of the RA patients. Several lines of evidence suggest that citrullinated antigens have direct involvement in the rheumatoid disease process. Anti-CCP antibodies precede the clinical development of synovitis by many years. B-cells are also very efficient antigen-presenting cells, and can contribute to T-cell activation. The important role of B-cells in the disease aetiology is supported by the recent success of B-cell depletion therapy using rituximab(Reference Edwards, Leandro and Cambridge120). The major effector cells in the pathogenesis of arthritis are synovial macrophages and fibroblasts. Activated macrophages are critical in RA, not only due to macrophage-derived cytokines (in particular TNF-α and IL-1) in the synovial compartments(Reference Firestein, varo-Gracia and Maki121), but also because of their localisation at strategic sites within the destructive pannus tissue. The variety and extent of macrophage-derived cytokines in RA and their widespread effect indicate that macrophages are local and systemic amplifiers of disease severity and perpetuation(Reference Kinne, Brauer and Stuhlmuller122). Among the many cell types present in the rheumatoid joint, the fibroblast-like synovial cell is prominent. It is now accepted that these cells are directly responsible for cartilage destruction, and also drive inflammation(Reference Kontoyiannis and Kollias123). There is evidence for proliferation and expression of inflammatory cytokines and chemokines by fibroblast-like synovial cells in inflamed synovia. Various studies have shown the presence and activity of regulatory T-cells in RA, both in peripheral blood and in the synovial compartment during active disease(Reference van Amelsfort, Jacobs and Bijlsma103, Reference de Kleer, Wedderburn and Taams109). Although in one of the RA studies anti-TNF interventions were shown to restore a compromised activity of regulatory T-cells(Reference Ehrenstein, Evans and Singh124), it seems that impaired T-cell regulation is a more prominent feature of multiple sclerosis and type 1 diabetes than RA.
Skin: atopic eczema and psoriasis
Atopic eczema (also called atopic dermatitis) and psoriasis are among the commonest inflammatory skin diseases. Both involve interactions between the immune system and the skin. Although broad acting immunosuppressive therapies are effective against both, very different pathogenetic mechanisms are involved.
Atopic eczema
Fifteen per cent of children will have eczema at some time during the first 12 years of their life(Reference Williams125). The clinical manifestations usually appear within the first year of life and often within a few weeks of birth. The rash of eczema is characteristically distributed (flexures) over ill-defined areas of intensely itchy redness containing an infiltrate of T lymphocytes and eosinophils. There is a clear genetic predisposition but expression of the phenotype is determined by environmental factors (among those suggested are lack of bacterial exposure providing an immune stimulus, intra-uterine exposure to allergens, poor fetal nutritional status). A major component in the pathogenesis of atopic eczema is the involvement of the immune system through a combination of immediate type (IgE-mediated) and T-cell-mediated immune hypersensitivities to environmental aeroallergens and to food allergens. Although the different types of hypersensitivity are demonstrable by prick test, intradermal injection or epicutaneous patch test challenge with a range of allergens, proving that the causal relevance of specific allergies is difficult and unreliable. The main effective therapies for atopic dermatis include topical steroids and systemic immuno-suppressives.
A fundamental component of the atopic state appears to be a dysregulation of the immune system such that T lymphocytes responding to atopic allergens differentiate towards the Th2 phenotype(Reference Krutmann and Grewe126, Reference Thepen, Langeveld-Wildschut and Bihari127). This may represent a failure of maturation of the fetal immune system, which is maintained during pregnancy with a bias towards Th2 responses(Reference Saito128). Signals from DC are thought to determine the differentiation pathway of Th cells. Microbial components including lipopolysaccharides signalling through innate receptors such as toll-like receptors are critical in this, as reflected by the hygiene hypothesis(Reference Strachan54, Reference von Mutius, Braun-Fahrlander and Schierl129–Reference Romagnani131). This proposes that early life exposure to microbes accelerates the maturation of the immune system away from the fetal Th2 bias and towards the adult Th1-biased differentiation pathway. Monocytes from atopic eczema sufferers release increased quantities of PGE2, which can drive T-cell differentiation towards the Th2 phenotype(Reference Chan, Kim and Henderson132, Reference Chan, Henderson and Li133).
Psoriasis
Two to three per cent of the population suffer from psoriasis; 10–15 % of these have it severely and require management by dermatologists. Fifty per cent of these will have significant joint symptoms and psoriatic arthritis. There is a genetic susceptibility and a number of genetic loci have been identified that may contribute to this susceptibility. There are associations with hyperlipidaemia and CHD(Reference Ena, Madeddu and Glorioso134, Reference Rocha-Pereira, Santos-Silva and Rebelo135) as well as CD(Reference Bernstein, Wajda and Blanchard136). Various factors are involved in the expression of the psoriatic phenotype: streptococcal infections; psychological stress; physical trauma to the skin. The rash of psoriasis is characterised by plaques of red scaly skin characteristically distributed over bony prominences. The pathophysiology involves an interaction between the immune system and the skin. There is an infiltrate of T lymphocytes (both CD4+ and CD8+) into the dermis, formation of clusters of neutrophils in the epidermis, involvement of two or three layers of the epidermis in proliferation and a greatly accelerated but incomplete differentiation. There are also proliferation and altered structure of the dermal capillaries, which become tortuous and dilated. Activation of the innate immune system by streptococcal products and, most likely, as yet unidentified factors, induces release of cytokines including IFN-α and -γ. The cellular source is unclear but could be plasmacytoid DC or other DC. This activates keratinocytes to proliferate and produce angiogenic factors that induce proliferation of dermal microvessels. The fundamental abnormality has not been identified, but the central feature is a failure of mechanisms of resolution of inflammation. The main treatment modalities include agents that modulate both keratinocyte proliferation and activation of T-cells. These include topical agents and systemic drugs. While many therapies have diverse effects on immune cells, cytokine production and/or cellular proliferation, it is unclear what the critical therapeutic target is. Retinoids, vitamin D analogues and glucocorticoids all have significant therapeutic efficacy and have in common action via the superfamily of ligand-activated nuclear transcription factors. The possibility is that nutritional intervention is suggested by the evidence of the involvement of eicosanoid mediators in psoriasis. Thus, there are reports that inhibition of leukotriene B4 (LTB4) with benoxaprofen was effective against psoriasis(Reference Thepen, Langeveld-Wildschut and Bihari127, Reference Kragballe and Herlin138).
The vascular wall: CVD
Medium-sized arteries, such as the coronary vessels, consist of three compartments termed the intima, media and adventitia, respectively. The intima is lined by a single continuous layer of endothelial cells. The endothelium forms the interface between the constituents of blood and the artery wall. Collectively, the cells of the endothelium have a mass of more than a kilogram and a surface area equivalent to that of a football pitch. Endothelial cells differ in their properties at different anatomical sites in the vascular tree. Such differences include variations in permeability and cytokine expression. Endothelial heterogeneity may be partially responsible for the characteristic regional distribution of atherosclerotic lesions described in human post-mortem studies. The endothelium appears to play a critical role in the regulation of vascular tone, and inhibiting leucocyte adhesion and platelet aggregation, through its release of mediators such as NO and prostacyclin(Reference Anggard139). NO is derived from l-arginine through the action of the constitutive form of the enzyme, endothelial NO synthase. It inhibits platelet aggregation and adhesion, modulates smooth muscle cell proliferation, attenuates the generation of endothelin and reduces leucocyte adhesion to the endothelium. The intima and media are separated from each other by a thin layer of elastic tissue called the internal elastic laminar. The media is composed of spirally arranged smooth muscle cells responsible for the generation of tone within the artery and determining the luminal diameter.
Atherogenesis
Atherosclerosis, or ‘hardening of arteries’ is the major cause of CVD. It is most often due to the formation of atheroma. Endothelial dysfunction is thought to presage atherosclerosis, and is characterised by altered endothelial function, enhanced adhesion molecule expression, increased leucocyte adhesion and impaired endothelium-dependent vasodilator responses(Reference Penn, Rangaswamy and Saidel140–Reference Kupatt, Zahler and Seligmann143). Although the endothelium appears to continue to elaborate NO, its biological activity appears to be compromised in the early phases of atherogenesis(Reference Ohara, Peterson and Harrison144). The latter is likely to be due in part to the interaction of NO with other molecular species such as the superoxide radical. These interactions neutralise the protective effects of NO and generate products, such as peroxynitrite that may be cytotoxic and pro-inflammatory. These changes in the properties of the endothelium have given rise to the concept of endothelial dysfunction. Endothelial dysfunction is associated with enhanced thrombosis and impaired fibrinolysis, and a number of risk factors appear to contribute to endothelial injury, including smoking, hypertension and hyperlipidaemia. The emergence of the ‘lipid oxidation’ hypothesis(Reference Steinberg, Parthasarathy and Carew145) provided yet another mechanism of endothelial injury, and an explanation for the formation of lipid-laden, macrophage-derived foam cells. Uptake of cholesterol by cells is normally mediated by the LDL receptor, and in the presence of high cellular levels of cholesterol, the LDL receptor is downregulated, limiting cholesterol accumulation. However, the uptake of oxidatively modified LDL is mediated by ‘scavenger receptors’ and this mechanism is not regulated by cellular cholesterol content. The putative protective effect of antioxidants, such as vitamin E, that inhibit LDL oxidation is explained by this hypothesis, although it is important to note that these effects remain contentious(Reference Miller, Pastor-Barriuso and Dalal146, Reference Bjelakovic, Nikolova and Gluud147).
Leucocytes become attached to the dysfunctional endothelium and subsequently accumulate within the subendothelial space(Reference Masuda and Ross148, Reference Masuda and Ross149). Macrophages are converted to lipid-laden foam cells within the artery wall, giving rise to a lesion termed the fatty streak. The initial adhesion event is mediated by pairs of adhesion molecules. The endothelial adhesion molecules have been shown to be upregulated early in atherogenesis.
The progression of the atherosclerotic plaque is complex process, and in its early stages of development may be reversed(Reference Stary150, Reference Stary151). The conversion of the fatty streak into a fibrous plaque necessitates the recruitment and proliferation of vascular smooth muscle cells(152). This process is driven by the synergistic interplay of several growth factors.
Fatal myocardial infarction is usually associated with thrombi and plaque fissuring. Unstable plaques are thought to be particularly prone to fissuring, and these are characterised by a large lipid pool, thin fibrous cap and large numbers of inflammatory cells(Reference Lendon, Davies and Born153). Activated macrophages within the plaque are a rich source of matrix metalloproteinases that have the ability to degrade extracellular matrix(Reference George154). They therefore have the potential to destabilise the plaque, leading to localised regions of de-endothelialisation that may subsequently lead to focal thrombosis and plaque rupture. In the later stages of its evolution, a plaque may be stabilised. Although such lesions may be sufficiently prominent to cause luminal narrowing, and hence angina, or claudication, they are less likely to rupture, and are therefore considered relatively safe.
The role of chronic inflammation in atherogenesis
Atherosclerosis bears many hallmarks of a chronic inflammatory disease, and every stage of its evolution is characterised by monocyte/macrophage and T-lymphocyte infiltration(Reference Katsuda, Boyd and Fligner155). The possible stimuli to this inflammatory process include oxidised LDL, homocysteine, free radicals generated from cigarette smoking, and infectious micro-organisms, such as Chlamydia pneumoniae. The T-cell infiltrates are predominantly CD4+ cells and clones derived from human lesions have been found to react to antigens derived from oxidised LDL, heat-shock proteins and micro-organisms(Reference Hansson156). The cytokine milieu within atherosclerotic lesions is thought to promote a Th1-dominated response associated with macrophage activation and the elaboration of IFN-γ and IL-1β(Reference Hansson156). Hence, if the original insult is not adequately neutralised, inflammation may persist, causing the local and systemic release of growth factors and cytokines. These can, in turn, lead to intimal thickening by stimulating smooth muscle cell migration, proliferation and extracellular matrix elaboration.
Inflammatory biomarkers and atherosclerosis
There has been an increasing interest in the use of inflammatory markers to estimate the risks of acute events in patients with established coronary disease. In part, the predictive value of these markers may be related to their ability to identify patients with vulnerable plaques, which are rich in activated leucocytes. CRP, IL-6 and circulating leucocyte count are elevated in patients with myocardial infarction(Reference Rifai and Ridker157). There have also been reports of a positive relationship between serum CRP and subclinical atherosclerosis(Reference Hulthe, Wikstrand and Fagerberg158), although analysis may be confounded because risk factors such as smoking habit, indices of adiposity, blood pressure, TAG and HDL are also associated with CRP concentrations. Adipose tissue may be an additional source of inflammatory cytokines that could stimulate CRP production(Reference Mohamed-Ali, Coppack and Stanner159). Other inflammatory conditions, such as RA and systemic lupus erythematosus, are associated with impaired endothelial function and increased coronary risk(Reference Vaudo, Marchesi and Gerli160, Reference Bruce, Urowitz and Gladman161).
Epidemiology of CVD
The Framingham Heart and Seven Countries Studies were key to identifying cigarette smoking, high serum cholesterol, physical inactivity, high blood pressure, the menopause and psychological factors as contributors to coronary disease. The importance of multiple, low-level risk factors has become clearly evident(Reference Kannel, D'Agostino and Sullivan162). Death rates from CVD have been falling in many developed countries, including most countries of Western Europe, since the early 1970s(Reference Unal, Critchley and Capewell163–166) and much of this reduction appears to be due to successful implementation of primary intervention due to dietary change, statin treatment, antihypertensive treatment and fall in smoking habit. The rate of reduction in CVD mortality differs among countries. For example, the fall in the UK is slower than that in Australia, Canada and Sweden. However, death rates from CVD have risen rapidly in many Eastern European countries. Poland is an exception to this because rates have fallen in association with a fall in the prevalence of smoking, reduced saturated fat and increased PUFA and fruit consumption(Reference Zatonski and Willett167). Besides national differences, there are very marked regional differences within countries(166, 168), which may reflect socio-economic differences. The incidence of CHD varies with race and ethnicity(Reference Frayn, Stanner and Stanner169). In the USA, black men and women have a higher risk of CHD than white men and women, respectively, while Hispanic groups have lower CHD rates. South Asians living in the UK have approximately 50 % higher rates of premature CHD death, and the difference is widening as rates do not appear to be falling as fast among this group as for the UK as a whole. This may relate to a higher prevalence of diabetes in this group.
Interventions
The landmark statin trials have demonstrated that cholesterol lowering reduces cardiovascular mortality and morbidity(Reference Baigent, Keech and Kearney170). The early benefits observed in some of the primary prevention trials have suggested that statins may be exerting effects other than those attributable to cholesterol lowering alone, so-called ‘pleiotropic effects’ including effects on inflammation, although this remains a matter of considerable debate. The role of low-dose aspirin treatment in the prevention of coronary disease is also well supported by trial data(Reference Baigent, Sudlow and Collins171). However, the mechanism for the protective effect may relate to the anti-platelet effects of aspirin, rather than an effect on inflammation. Although trials of antibiotic treatment have generally not shown a benefit in preventing coronary disease(Reference Taylor-Robinson and Boman172), it has been argued that diet regimens used to date may not eliminate the carriage of organisms by peripheral monocytes. There have been few studies describing the effects of dietary constituents on inflammation in healthy subjects or patients with CHD. Fish oil supplements, providing very long-chain n-3 PUFA (VLC n-3 PUFA) alter the composition of inflammatory cells resulting in decreased production of inflammatory eicosanoids and cytokines(Reference Calder173), alter cellular composition of human plaques, making them appear more stable(Reference Thies, Garry and Yaqoob174), and reduce mortality from sudden death in secondary prevention(175, Reference Marchioli, Barzi and Bomba176). Meta-analyses support lowered cardiovascular mortality with increased VLC n-3 PUFA intake(Reference Bucher, Hengstler and Schindler177, Reference Studer, Briel and Leimenstoll178).
Adipose tissue: obesity
Adipose-derived mediators
The prevalence of obesity (defined as a BMI >30 kg/mReference Elewaut, DiDonato and Kim2) is rising throughout the world, and obesity is occurring at a younger age. In the US and most Western countries, obesity has an incidence rate above 20 %. This is a major public health issue because of the direct effect of obesity on the risk of developing type 2 diabetes, CVD, hypertension, stroke, abnormal blood lipids, arthritis, asthma and cancer(179). Until very recently, the role of adipose tissue has been thought to be passive and adipocytes were considered as little more than fat stores. However, it is now clear that this is far from the case and that adipose tissue is an important endocrine organ(Reference Hutley and Prins180). There is increased understanding that, depending on the nutritional state, the profile of hormones released from adipocytes can change from being beneficial to being detrimental(Reference Fantuzzi181). The adipocyte plays a major role in the inflammatory response and releases a cocktail of inflammatory mediators and signalling molecules(Reference Halaas, Gajiwala and Maffei182, Reference Montague, Farooqi and Whitehead183).
Four findings ignited the interest in the adipocyte as an endocrine organ. Firstly, the discovery of leptin in 1995 changed the way the adipocyte was viewed; the discovery that leptin has a role in signalling the status of adipose tissue to the brain was the first description of a feedback loop involved in body weight control(Reference Halaas, Gajiwala and Maffei182). Although its role in human subjects is more complicated, the importance of leptin as a hormone involved in energy balance has been exemplified by the case studies of families with leptin deficiency; the children are obese and lose weight with leptin therapy(Reference Montague, Farooqi and Whitehead183). As human subjects gain weight, they seem to develop leptin resistance. This may be mediated by suppressor of cytokine signalling, which increases in the hypothalamus in tandem with leptin. This blocks the central effect of leptin(Reference Greenberg and Obin184). Leptin has important effects on peripheral metabolism, which have been demonstrated through lipodystrophy models. Leptin has been shown to have a role to play in insulin sensitivity and TAG clearance from the circulation. This observation in evolutionary terms may have allowed weight gain in times of plenty without gross metabolic change.
Secondly, the discovery that adipocytes release TNF-α also changed the view of adipocytes(Reference Hotamisligil, Peraldi and Budavari185). In fact, mRNA expression studies show that adipocytes can synthesise TNF-α and several other cytokines, notably IL-1β and IL-6 among an ever-increasing list. Circulating TNF-α and IL-6 concentrations are mildly elevated in obesity. Most studies suggest increased TNF-α mRNA expression or secretion in vitro in adipose tissue from obese subjects. The factors regulating cytokine release within adipose tissue appear to include not only typical inflammatory stimuli such as lipopolysaccaride, but also the size of the fat cells per se and catecholamines. The effects of cytokines within adipose tissue include some actions that might be characterised as metabolic. TNF-α and IL-6 inhibit lipoprotein lipase, and TNF-α additionally stimulates hormone-sensitive lipase. TNF-α also downregulates insulin-stimulated glucose uptake via effects on GLUT-4, insulin receptor autophosphorylation and insulin receptor substrate-1. All these effects will oppose lipid accumulation within adipocyte(Reference Coppack186). Other effects appear more trophic, and include the induction of apoptosis, regulation of cell size and induction of de-differentiation (the latter involving reduced PPAR-γ). Cytokines are important stimulators and repressors of other cytokines. In addition, cytokines appear to modulate other regulatory systems. Examples of the latter include effects on leptin secretion (probably stimulation followed by inhibition) and reduction in β3-adrenoceptor expression. There seems to be no clear agreement as to which cytokines derived from adipose tissue act as remote regulators, i.e. hormones. Leptin, which is structurally a cytokine, is also a hormone. IL-6 appears to be released systemically by adipose tissue, but TNF-α is probably not. Both leptin and IL-6 appear to act on the hypothalamus, IL-6 acts on the liver, while leptin may have actions on the pancreas(Reference Coppack186).
Thirdly, the discovery of adiponectin, a protein which is specifically and very highly expressed in adipose tissue(Reference Gil-Campos, Cañete and Gil187). This hormone enhances insulin sensitivity in muscle and liver and increases fatty acid oxidation in several tissues, including muscle fibres. It also decreases serum fatty acid, glucose and TAG concentrations: if normal, lean mice are given injections of adiponectin in conjunction with a meal high in fat and sugar, the normal postprandial increases in plasma glucose and TAG concentrations are smaller as the result of an increased rate of clearance from the blood rather than a reduced rate of absorption from the gut. By contrast, if insulin-resistant mice are treated with physiologic concentrations of adiponectin, glucose tolerance is improved and insulin resistance is reduced. In human subjects, plasma adiponectin concentrations fall with increasing obesity and this effect is greater in men than women. Reduced adiponectin concentrations correlate with insulin resistance and hyperinsulinaemia. In addition, several polymorphisms of the adiponectin gene (APM1, mapped to chromosome 3q27) have been identified that are associated with reduced plasma adiponectin concentration and that increase the risk of type 2 diabetes, insulin resistance or the metabolic syndrome. Interestingly, adiponectin appears to be implicated in the development of atherosclerosis. Adiponectin concentrations are reduced in patients with CHD, and adiponectin inhibits TNF-α-induced expression of adhesion molecules and the transformation of macrophages to foam cells, both of which are key components of atherogenesis. In mice deficient in apo E (and thus susceptible to atherosclerosis), treatment with human adiponectin inhibits lesion formation in the aortic sinus by 30 % compared with that in untreated control animals(Reference Fantuzzi181, Reference Greenberg and Obin184). The central importance of adiponectin is exemplified by the observation that it inhibits NF-κB and reduces the proinflammatory pathway.
Fourth is the observation that adipose tissue is not homogeneous. Macrophage numbers in adipose tissue increase with obesity, where they apparently function to scavenge moribund adipocytes, which increase dramatically with obesity. Macrophages are responsible for most of the cytokine production in adipose tissue, especially in obesity. In fact, adipose tissue macrophages are responsible for perhaps as much as 50 % of adipose tissue TNF-α production(Reference Greenberg and Obin184). In obesity, adipose tissue contains an increased number of resident macrophages so that, in some circumstances, macrophages can constitute up to 40 % of the cell population within an adipose tissue depot.
An integrated view of inflammation and obesity
The issues surrounding weight gain and inflammation cannot be seen in isolation, but need to be viewed alongside theories about variation in adipocyte differentiation, appetite regulation and control of appetite. Recent evidence has suggested that the formation of hypertrophic fat cells in environments of energy excess may be due to a genetically determined differentiation limit of stem cells to adipocytes or metabolic feedback controlling differentiation or more likely a mixture of the two. If there are limited numbers of adipocytes in an environment of energy excess, this will cause excessive lipid filling in the adipocyte and lead to hypertrophic adipocytes that are pro-inflammatory. The key issue at the beginning of the inflammatory process seems to be an excess energy intake. Many of the processes that follow excess energy intake may acutely be an advantage, but an environment of chronic excess energy intake and decreased energy output will work against human health. As the adipocyte stores TAG, there seems to be an increase in cytokine release. Of particular importance is the suppression of adiponectin and the increase in TNF-α. It is possible that TNF-α can decrease proliferation and differentiation of adipocytes. The control mechanisms for adipocyte proliferation and differentiation are complex, but examination of the transcriptional and extracellular signals necessary for stem cell differentiation into a preadipocyte, and from the preadipocyte into a mature fat cell, is being elucidated. Regulation of adipocyte proliferation and differentiation involves growth arrest and the coordinate activation/inactivation of nuclear transcription factors that regulate the expression of genes necessary for lipid storage and insulin sensitivity. ADD/SREB-1, C/EBP-a, -b and -d and PPAR-γ are a few of the transcription factors known to be involved. These transcription factors are regulated in response to extracellular signals, such as PG, cytokines, and hormones including corticosteroids and insulin(Reference Hausman, DiGirolamo and Bartness188). Defects in any one of these steps are potentially important in the failure of proliferation or differentiation of adipocytes. This will limit the number of adipocytes coping with the excess energy intake. This will have several effects.
(i) Effects on recruitement of monocytes into adipose tissue. As the lipid content of adipose tissue increases, the adipocytes increase leptin output and this has the effect of increasing expression of adhesion molecules such as ICAM-1 and platelet-endothelial cell adhesion molecule-1 in endothelial cells inducing adhesion and transmigration of monocyte/macrophages. The transmigration of macrophages is further enhanced by monocyte chemoattractant protein-1. The result is a tissue that produces the low-grade systemic inflammation seen in obesity(Reference Neels and Olefsky189).
(ii) Effects on apoptosis and TNF-α. In mature white adipocytes, TNF-α is reported to regulate cell size, the suggestion being that as the cells get bigger they produce more TNF-α, which in turn initiates changes to limit adipocyte size or to induce apoptosis. This is possibly coupled with increasing local hypoxia. The net result is recruitment of greater numbers of macrophages that surround the apoptotic cell(Reference Coppack186, Reference Neels and Olefsky189).
(iii) Autocrine effects on adipokines. The increase in TNF-α has been shown to have several autocrine effects including reduced expression of the GLUT4 glucose transporter, increased hormone-sensitive lipase activity leading to increased lipolysis, decreased lipoprotein lipase activity, reduced insulin signalling and decreased C/EBP and PPAR-γ activity, so creating an insulin insensitive adipocyte with a high output of NEFA(Reference Coppack186).
(iv) Effects on peripheral tissue. From the earlier adipocyte hypertrophy, increased insulin resistance and increased fatty acid output result and excess dietary fat may be shunted to the liver, skeletal muscle and the β-cell. Importantly, if fat oxidation cannot be increased to compensate for the increased influx of lipid within these tissues, then intracellular accumulation of lipids will occur. The oxidation of fat is mediated by activating AMP-activated protein kinase (AMPK). It is known that adiponectin activates AMPK as does leptin. So, in normal physiology, leptin and adiponectin signal transduction involves the phosphorylation of STAT pathways, but how this translates into the observed changes in lipid metabolism is not clear. One likely mechanism is via increased phosphorylation activation of AMPK, which phosphorylates acetyl CoA carboxylase and malonyl CoA decarboxylase. Phosphorylation inactivates carboxylase, but activates malonyl CoA decarboxylase. Because carboxylase catalyses malonyl CoA formation, and malonyl CoA decarboxylase catalyses its decarboxylation, the net effect of AMPK activation on these target enzymes is to lower malonyl CoA concentration. Malonyl CoA is the first committed step in lipogenesis and a powerful inhibitor of carnitine palmityl transferase-1-mediated fatty acid oxidation. The combination of an increase in fatty acid oxidation and a decrease in fatty acid synthesis could account for the reduction in the lipid content of a cell. At present, however, we do not understand how leptin activates AMPK. In the inflammatory state there is low adiponectin and leptin resistance and therefore low activity of AMPK, and reduced oxidation of fat and the storage of TAG in cells. It is likely that it is products generated from TAG such as unoxidised palmitoyl-CoA that lead to insulin resistance and cell apotosis. Also diacylglycerol has been shown to have effects on protein kinase C; this can in turn affect the insulin signal by causing serine phosphorylation rather than tyrosine phosphorylation on IRS-1 so reducing the insulin signal. This theory is supported by the recent observation that there is a direct relationship between hepatocyte and myocyte lipid content and insulin sensitivity. Interestingly, IL-6 can also affect steroid hormone conversion, changing the balance of sex hormones, which is well known to regulate adipose tissue distribution(Reference Unger190–Reference McGarry192).
The release of other cytokines (e.g. plasminogen activation inhibitor 1 (PAI-1)) from the adipocyte can have dramatic effects on risk of disease. There is evidence for an association between increased PAI-1 levels and myocardial infarction. Interestingly, PAI-1 has been shown to predict myocardial events in univariate analysis, but the predictive power was not affected by adjustement for inflammatory parameters and disappeared after adjustment for BMI, TAG and HDL cholesterol. This suggests that the metabolic syndrome is a precursor to high plasma PAI-1 levels in patients prone to atherosclerosis(Reference Alessi, Lijnen and Bastelica193).
Effect of weight loss on inflammatory markers
The induction of a negative energy balance has profound effects on inflammatory markers in obesity: weight loss with diet and exercise has been shown to decrease circulating IL-6 and TNF-α concentrations in obese subjects(Reference Bougoulia, Triantos and Koliakos194, Reference Cancello and Clement195). Serum CRP concentration fell 4 months after bariatric surgery but remained significantly elevated relative to non-obese controls(Reference Zagorski, Papa and Chung196). Very recently, there has been a description of a fall in proinflammatory cytokines and macrophage numbers in white adipose tissue following weight loss(Reference Cancello and Clement195). A recent study on obese children has demonstrated falls in plasma TNF-α and CRP concentrations with weight loss(Reference Reinehr, Stoffel-Wagner and Roth197). Interestingly, liposuction has very little effect on inflammatory markers(Reference Hansen, Torquati and Abumrad198). The success of lifestyle intervention has been demonstrated with the American and Finnish Diabetes Prevention trials, which both show long-term benefit in decreasing the risk of type 2 diabetes(Reference Knowler, Barrett-Connor and Fowler199, Reference Tuomilehto, Lindstrom and Eriksson200).
Common features of inflammatory processes that may be suitable targets for nutritional intervention
Although inflammation-induced tissue damage occurs in an organ-specific manner (gut, lungs, joints, skin, blood vessel wall, adipose tissue) in different diseases or conditions, there is some commonality among the responses seen in the different organs (summarised in Table 1). In general, the inflammatory response observed is normal but it occurs in the wrong context, which relates to inappropriate barrier function (epithelial or endothelial), inappropriate triggering (i.e. a response to a normally benign stimulus equivalent to a loss of tolerance), lack of downregulation to control the response and tissue destruction with a loss of function. In some cases, the inflammation is the result of tissue damage caused by endogenous molecules such as necrotic cell debris or oxidised LDL. In other cases, it is the result of exogenous triggers such as allergens or microbes. The involvement of different triggers is also reflected in the distinct associations with polymorphisms in receptors involved in ‘pattern’ recognition such as NOD2 in CD or with other molecules involved in specific adaptive immune responses such as HLA-DR subtypes in UC and RA (Table 1). However, although trigger, localisation and resulting clinical symptoms are different, many of the processes, cells and molecules involved in the actual inflammatory response are remarkably similar (Table 1). Most, if not all, of the chronic inflammatory diseases considered here are characterised by overproduction of cytokines (TNF-α, IL-1β, IL-6, IFN-γ), chemokines (IL-8, MCP-1), eicosanoids (PGE2, four-series LTs) and matrix metalloproteinases. Elevated levels of these mediators act to amplify the inflammatory process (e.g. by attracting further inflammatory cells to the site) and contribute to tissue destruction and to the clinical symptoms observed. Many of these mediators are positively regulated through NF-κB (Fig. 3) and some are negatively regulated through PPAR and liver X receptors. Entry of inflammatory cells to sites of inflammatory activity is facilitated by upregulation of adhesion molecules on the endothelium, a process that is promoted by inflammatory cytokines and by a range of inflammatory triggers, frequently acting through NF-κB. The continuous process of tissue injury, healing and repair, due to the release of cytokines, chemokines and growth factors by infiltrating inflammatory cells as well as resident tissue cells, results in tissue remodelling. Local structural cells, such as fibroblasts, epithelial cells and smooth muscle cells can play a role in amplification of the inflammatory response and to a large extent determine the clinical appearance of the disease.
Table 1 Summary of the disease states discussed and an overview of their key inflammatory components

COPD, chronic obstructive pulmonary disease; HLA, human leucocyte antigen; NOD, nucleotide-binding oligomerisation domain; IBD, inflammatory bowel disease; Th, helper T-cell; PMN, polymorphonuclear cells; IFN, interferon; MMP, matrix metalloproteinase; cysLT, cysteinyl leukotriene; LT, leukotriene; MCP, monocyte chemoattractant protein; CDAI, Crohn's disease activity index; CRP, C-reactive protein; CAI, colitis activity index; FEV1, forced expiratory volume at 1 s.
Fig. 3 NF-κB signalling pathway.
It is noteworthy that in many cases, at some point, the tissue damage appears to result in a loss of barrier function and exposure of hitherto sequestered antigens of either the host or of its microbiota. This can result in a loss of tolerance and sustained specific responses to such antigens. This can involve quite distinct antigens such as the gut microflora in CD and UC, fillagrin in eczema, and cartilage in RA. In all these cases, these responses seem to result from loss of barrier function and once initiated help to sustain the chronic inflammatory condition.
Why resolution of inflammation is absent or abnormal in so many pathophysiological processes remains largely unknown, although there are a number of mechanisms that may be considered.
(i) Persistent insult (i.e. chronic infection or continued exposure to initiating allergens) provides continued pro-inflammatory stimuli.
(ii) The inflammatory response results in tissue damage and loss of barrier function resulting in exposure of sequestered antigens and loss of tolerance to autoantigens or components of the microbiota, which then provide a trigger to drive prolonged inflammation.
(iii) Local (over)production of survival factors such as IL-5, granulocyte–macrophage colony stimulating factor and IL-1β results in prolonged survival and activity of granulocytes.
(iv) Defective or reduced phagocytosis impairing clearance of cellular debris. This results in granulocyte necrosis and the release of reactive oxygen and nitrogen species as well as the activation of macrophages leading to sustained production of pro-inflammatory cytokines.
(v) Deficiency in immunological negative feedback mechanisms, which leads to loss of inflammatory control. This could be at the level of regulatory T-cell dysfunction (no release of anti-inflammatory cytokines), at the level of intracellular negative feedback mechanisms in immune cells or impaired macrophage suppresser activity (no release of regulatory proteins).
Although the relative importance may differ, these mechanisms seem to contribute to most of the conditions described here. Over time, chronic inflammation also results in a gradual overload with reactive oxidant species, which when insufficiently countered by the gradually weakened anti-oxidative defence systems results in a shift in redox balance. This in turn leads to uncontrolled activation of the redox-sensitive proinflammatory transcription factor NF-κB, continued production of reactive oxygen and nitrogen species and supports chronic inflammation (Fig. 4).
Fig. 4 Representation of the interaction between oxidant stress and inflammation. IκB, inhibitory subunit of NF-κB; IL, interleukin; NF-κB, nuclear factor κ B; PG, prostaglandin; TNF, tumor necrosis factor.
The foregoing considerations suggest a number of potential targets for nutritional intervention, which are common across the various inflammatory pathologies considered here. Among these targets are common mediators, including ROS, arachidonic acid-derived lipid mediators (PG, LT), chemokines (IL-8, MCP-1), inflammatory cytokines (TNF-α, IL-1, IL-6, IFN-γ, etc.) and matrix metalloproteinases, and common intracellular signalling factors including the transcription factors NF-κB and PPAR-γ and their upstream regulators.
Nutrition and inflammatory processes
Fatty acids
Description, sources, normal intakes and roles
The general structure of a fatty acid is a hydocarbon (acyl) chain of varying length with a carboxyl group at one end and a methyl group at the other. The carboxyl group is reactive and readily forms ester links with alcohol groups for example those on glycerol or cholesterol. The most abundant fatty acids have straight chains of an even number of carbon atoms. Fatty acids containing double bonds are referred to as unsaturated fatty acids; a fatty acid containing two or more double bonds is called a PUFA. Most common unsaturated fatty acids contain cis rather than trans double bonds. However, trans double bonds do occur in ruminant fats (e.g. cow's milk), in plant lipids and in some seed oils. In some PUFA, the double bonds are not separated by a methylene (–CH2–) group, but are conjugated. The systematic name for a fatty acid is derived from the number of carbon atoms in the acyl chain, the number of double bonds, the position of the double bonds (counting the carboxyl carbon as carbon 1) and their configuration (cis or trans). An alternative shorthand notation for fatty acids is frequently used. This relies upon identifying the number of carbon atoms in the chain, the number of double bonds and the position of the double bond closest to the methyl terminus of the acyl chain (the methyl carbon is known as the n-carbon). Thus, cis 9, cis 12-octadecadienoic acid is notated as 18 : 2n-6. In addition to these nomenclatures, fatty acids are often described by a common name(Reference Calder, Burdge, Nicolaou and Kokotos201). Human subjects cannot synthesise the simplest n-6 and n-3 PUFA (linoleic acid (18 : 2n-6) and α-linolenic acid (18 : 3n-3)) and so these are regarded as essential fatty acids.
Fatty acids in fats, oils and foodstuffs are mainly esterified to glycerol, as TAG, although some are present as esterified components of phospholipids, glycolipids and other lipids. With regard to inflammatory processes, most attention has been paid to the effects of n?6 and n-3 PUFA(Reference Calder173, Reference Calder202), although other fatty acid classes including saturated and trans fatty acids may also influence inflammatory processes. Important dietary sources of the n-6 PUFA linoleic acid include vegetable oils (e.g. maize, sunflower, safflower and soyabean oils) and products made from those oils (e.g. margarines). Oils like soyabean also contain α-linolenic acid; other good sources of this fatty acid include some nuts, flaxseeds and flaxseed (linseed) oil. Some seed oils contain moderate to high proportions of relatively unusual fatty acids (e.g. γ-linolenic acid (18 : 3n-6) in borage (starflower) and evening primrose oils). Meat is an important source of the VLC n-6 fatty acid, arachidonic acid (20 : 4n-6). Fish can be classified into lean fish that store lipid as TAG in the liver (e.g. cod) or ‘fatty’ (‘oily’) fish that store lipid as TAG in the flesh (e.g. mackerel, herring, salmon, tuna, sardines). The oil obtained from fatty fish flesh or lean fish livers is termed ‘fish oil’ and it has the distinctive characteristic of being rich in the VLC n-3 PUFA eicosapentaenoic acid (EPA; 20 : 5n-3) and DHA (22 : 6n-3).
There are large differences in fat intake among countries with average intakes among adults varying from < 20 g/d in some developing countries to >100 g/d in some developed countries. The mix of fatty acids consumed also varies in accordance with the fatty acid compositions of the fats and oils used in food preparation and of the foodstuffs eaten. Average fat consumption has changed over time and continues to do so. In many developing countries fat intake is increasing, while in developed countries fat intake has tended to decline over the last 40 years or so.
The type of fat consumed has also changed over time, meaning that the fatty acid composition of the human diet has changed. The latest figures for the UK indicate that adult males consume an average of 85 g fat/d and adult females 60 g/d (both approx. 35 % of dietary energy). The main PUFA in the diet is linoleic acid followed by α-linolenic acid. On average, adult men in the UK consume about 12 and 1·9 g linoleic and α-linolenic acids, respectively, per day. Adult women in the UK consume about 8·7 and 1·4 g linoleic and α-linolenic acids, respectively, per day. Hulshof et al. (Reference Hulshof, van Erp-Baart and Anttolainen203) provided details of intakes of fatty acids among 14 Western European countries, while Burdge & Calder(Reference Burdge and Calder204) included some data from North America and Australia. Longer-chain PUFA are consumed in lower amounts than linoleic and α-linolenic acids. Estimates of the intake of arachidonic acid in Western populations vary between 50 and 300 mg/d for adults(Reference Calder, Burdge, Nicolaou and Kokotos201). In the absence of fatty fish or fish oil consumption, α-linolenic acid is by far the principal dietary n-3 PUFA. Average intake of the VLC n-3 PUFA in the UK and in other Western countries where oily fish consumption is not the norm is estimated at < 250 mg per day(Reference Calder, Burdge, Nicolaou and Kokotos201).
Fatty acids are transported in the bloodstream largely in esterified form as components of lipoproteins, although albumin-bound NEFA also circulate(Reference Frayn205). Fatty acids have many diverse functions in cells; their principal roles are as energy sources and as membrane constituents(Reference Calder, Burdge, Nicolaou and Kokotos201). Many types of fatty acid can fill these roles. Certain fatty acids have additional, specific roles: arachidonic, dihomo-γ-linolenic (20 : 3n-6) and EPAs act as precursors for eicosanoid synthesis; myristic and palmitic acids may be covalently attached to certain proteins and this is believed to play an important role in the functioning of these proteins; fatty acids are able to influence intracellular signalling processes; fatty acids are able to influence transcription factor activity and gene expression. Through these different actions, fatty acids are able to influence cellular functions and so affect physiological responses, including inflammation.
Effects on inflammatory processes and mechanisms of action
Arachidonic acid, arachidonic acid-derived eicosanoids and inflammation
The key link between fatty acids and inflammation is that eicosanoids that act as mediators and regulators of inflammation are generated from 20-carbon PUFA. Because inflammatory cells typically contain a high proportion of the n-6 PUFA arachidonic acid and low proportions of other 20-carbon PUFA, arachidonic acid is usually the major substrate for eicosanoid synthesis. Eicosanoids, which include PG, thromboxanes, LT and other oxidised derivatives, are generated from arachidonic acid by reactions catalysed by cyclo-oxygenase (COX) and lipoxygenase (LOX) enzymes (Fig. 5). There are at least two COX enzymes and several LOX enzymes that are expressed in different cell types, according to different conditions and which between them produce a range of mediators involved in modulating the intensity and duration of inflammatory responses(Reference Lewis, Austen and Soberman206, Reference Tilley, Coffman and Koller207). These mediators have cell- and stimulus-specific sources and frequently have opposing effects. Thus, the overall physiological (or pathophysiological) outcome will depend upon the cells present, the nature of the stimulus, the timing of eicosanoid generation, the concentrations of different eicosanoids generated and the sensitivity of target cells and tissues to the eicosanoids generated.
Fig. 5 Eicosanoid biosynthesis from arachidonic acid. COX, cyclo-oxygenase; HETE, hydroxyeicosatetraenoic acid; HPETE, hydroperoxyeicosatetraenoic acid; LOX, lipoxygenase; LT, leukotriene; PG, prostaglandin; TX, thromboxane.
The amount of arachidonic acid in inflammatory cells can be increased by including it in the diet(Reference Calder173) and may also be influenced by the dietary intake of its precursor, linoleic acid, although the range of linoleic acid intake over which this relationship occurs has not been defined for human subjects. The role of arachidonic acid as a precursor for the synthesis of eicosanoids indicates the potential for dietary n-6 PUFA (linoleic or arachidonic acids) to influence inflammatory processes. However, this has been little investigated in human settings. Supplementation of the diet of healthy young male subjects with 1·5 g arachidonic acid/d for 7 weeks resulted in a marked increase in the production of PGE2 and LTB4 by endotoxin-stimulated mononuclear cells(Reference Kelley, Taylor and Nelson208). However, production of TNF-α, IL-1β and IL-6 by the latter was not significantly altered. Thus, increased arachidonic acid intake may result in changes indicative of selectively increased inflammation or inflammatory responses in human subjects. Supplementation of the diet of healthy elderly subjects with arachidonic acid (0·7 g/d in addition to a habitual intake of about 0·15 g/d) for 12 weeks did not affect endotoxin-stimulated production of TNF-α, IL-1β or IL-6 by mononuclear cells, did not alter superoxide production by neutrophils or monocytes and did not alter plasma-soluble adhesion molecule concentrations(Reference Thies, Miles and Nebe-von-Caron209).
Very long-chain n-3 PUFA and inflammatory eicosanoid production
Increased consumption VLC n-3 PUFA such as EPA and DHA, usually given as fish oil in experimemtal settings, results in increased proportions of those fatty acids in inflammatory cell phospholipids (see Calder(Reference Calder173) for references). This occurs in a dose–response fashion and is partly at the expense of arachidonic acid(Reference Healy, Wallace and Miles210, Reference Rees, Miles and Banerjee211). Since there is less substrate available for the synthesis of eicosanoids from arachidonic acid, fish oil supplementation of the human diet has been shown to result in decreased production of these mediators by inflammatory cells(Reference Calder173). Although most studies have used fish oil, Kelley et al. (Reference Kelley, Taylor and Nelson212) demonstrated that 6 g DHA/d resulted in decreased production of PGE2 and LTB4 by endotoxin-stimulated mononuclear cells.
EPA is also able to act as a substrate for both COX and 5-LOX, giving rise to eicosanoids with a slightly different structure to those formed from arachidonic acid. Thus, fish oil supplementation of the human diet has been shown to result in increased production of alternative eicosanoids (see Calder(Reference Calder173) for references). The functional significance of this is that the mediators formed from EPA are believed to be less potent than those formed from arachidonic acid (see Calder(Reference Calder173) for references).
In addition to VLC n-3 PUFA modulating the generation of eicosanoids from arachidonic acid and to EPA acting as substrate for the generation of alternative eicosanoids, recent studies have identified a novel group of mediators, termed E- and D-series resolvins, formed from EPA and DHA, respectively, by the sequential actions of COX-2 and LOX enzymes, which appear to exert anti-inflammatory and inflammation resolving actions (see Serhan et al. (Reference Serhan, Arita and Hong213) and Serhan(Reference Serhan214) for reviews).
Anti-inflammatory effects of very long-chain n-3 PUFA other than altered eicosanoid production
A number of dietary supplementation studies that used between 3·1 and 14·4 g EPA+DHA/d have demonstrated a time-dependent decrease in chemotaxis of human neutrophils and monocytes towards various chemoattractants; both the distance of cell migration and the number of cells migrating were decreased (see Calder(Reference Calder173) for references).
Cell culture and animal feeding studies report decreased expression of some adhesion molecules on the surface of monocytes, macrophages or endothelial cells following exposure to VLC n-3 PUFA (see Calder(Reference Calder173) for references). Supplementing the diet of healthy human subjects with fish oil providing about 1·5 g EPA+DHA/d resulted in a lower level of expression of ICAM-1 on the surface of blood monocytes stimulated ex vivo with IFN-γ(Reference Hughes, Pinder and Piper215). Dietary fish oil providing 1·1 g EPA+DHA/d was found to decrease circulating levels of soluble VCAM-1 in elderly subjects(Reference Miles, Thies and Wallace216), but it is not clear whether this represents decreased surface expression of VCAM-1.
Supplementation studies providing 3·1–8·4 g EPA+DHA/d have reported decreases in the production of ROS (superoxide and/or hydrogen peroxide) by stimulated human neutrophils and monocytes (see Calder(Reference Calder173) for references). A recent study has reported the dose–response relationship between EPA intake and decreased superoxide production by human neutrophils and identified that cells from older subjects seem to be more sensitive to EPA than those from young subjects(Reference Rees, Miles and Banerjee211).
Cell culture studies demonstrate that EPA and DHA can inhibit the production of inflammatory cytokines (TNF-α, IL-1β, IL-6, IL-8) by monocytes and endothelial cells (see Calder(Reference Calder173)). Fish oil feeding decreased ex vivo production of TNF-α, IL-1β and IL-6 by rodent macrophages (see Calder(Reference Calder173)). Supplementation of the diet of healthy human volunteers with fish oil providing more than 2 g EPA+DHA/d has been shown to decrease production of TNF, or IL-1 or IL-6 by mononuclear cells in some studies (see Calder(Reference Calder173)). Caughey et al. (Reference Caughey, Mantzioris and Gibson217) reported a significant inverse correlation between the EPA content of mononuclear cells and the ability of those cells to produce TNF-α and IL-1β in response to endotoxin. Kelley et al. (Reference Kelley, Taylor and Nelson212) showed that 6 g DHA/d for 12 weeks resulted in decreased production of TNF-α and IL-1β by endotoxin-stimulated mononuclear cells. Both EPA and DHA (4 g/d for 6 weeks) decreased plasma TNF-α concentrations in type 2 diabetics, although DHA was more potent(Reference Mori, Woodman and Burke218). It should be noted that there are also several studies that fail to show effects of dietary long-chain n-3 PUFA on production of inflammatory cytokines in human subjects (see Calder(Reference Calder173, Reference Calder202) for references and discussion).
α-Linolenic acid and inflammatory processes
Increased consumption of α-linolenic acid results in an increased content of EPA and docosapentaenoic acid in the membranes of inflammatory cells (see Burdge & Calder(Reference Burdge and Calder219) for references) in parallel with a decreased content of arachidonic acid. This might be expected to affect inflammation. A recent study has reported an inverse association between α-linolenic acid intake and plasma concentrations of IL-6 and sVCAM-1 but not of CRP, soluble TNF receptor-2, se-selectin or sICAM-1 in samples taken from the Nurses Health Study 1 cohort(Reference Lopez-Garcia, Schulze and Fung220). After adjustment for age, BMI, physical activity, smoking status, alcohol consumption and intakes of linoleic acid and SFA, there were significant negative associations between α-linolenic acid intake and plasma CRP, IL-6 and E-selectin concentrations(Reference Lopez-Garcia, Schulze and Fung220). These observations would suggest that increasing α-linolenic acid intake would result in decreased inflammation. Caughey et al. (Reference Caughey, Mantzioris and Gibson217) reported that 13·7 g/d α-linolenic acid for 4 weeks resulted in a decrease in ex vivo production of PGE2, TNF-α and IL-1β by endotoxin-stimulated mononuclear cells by 33, 27 and 30 %, respectively. By comparison, fish oil providing 2·7 g/d EPA+DHA decreased the production of these inflammatory mediators by 55, 70 and 78 %, respectively(Reference Caughey, Mantzioris and Gibson217). Thus, on a g/d basis, VLC n-3 PUFA are about eight to fourteen times more potent than α-linolenic acid with respect to this outcome in healthy subjects. In contrast to the observations of Caughey et al. (Reference Caughey, Mantzioris and Gibson217), several studies using lower intakes of α-linolenic acid (2–9·5 g/d) did not find effects on neutrophil chemotaxis, neutrophil respiratory burst, monocyte respiratory burst, TNF-α, IL-1β and IL-6 production by endotoxin-stimulated mononuclear cells, all studied ex vivo, or on soluble adhesion molecule concentrations (see Burdge & Calder(Reference Burdge and Calder219) for references). Furthermore, a study by Rallidis et al. (Reference Rallidis, Paschos and Papaioannou221) reported no effect of 8 g/d α-linolenic acid on sICAM-1 or SE-selectin concentrations and a similar decrease in sVCAM-1 concentration in both α-linolenic acid and control groups. Likewise, Bemelmans et al. (Reference Bemelmans, Lefrandt and Feskens222) found no effect of 6·3 g/d α-linolenic acid on sICAM-1 concentration at 1 and 2 years of intervention. Taken together, these data suggest that increasing α-linolenic acid intake to >9 g/d is required in order for marked anti-inflammatory effects to be seen. Even then, the effects will be much more modest than those exerted by VLC n-3 PUFA(Reference Caughey, Mantzioris and Gibson217). However, both Rallidis et al. (Reference Rallidis, Paschos and Liakos223) and Bemelmans et al. (Reference Bemelmans, Lefrandt and Feskens222) did find a significant decrease in CRP concentration, suggesting that this may be a marker that is more sensitive to the intakes of α-linolenic acid, which do not affect soluble adhesion molecule or cytokine concentrations or ex vivo inflammatory cell responses. One study using a very high intake of α-linolenic acid (approx. 17·5 g/d) reported significant decreases in both CRP and soluble adhesion molecule concentrations(Reference Zhao, Etherton and Martin224). Interestingly, the authors found that the changes in these inflammatory markers were significantly related to changes in serum concentrations of EPA or EPA plus docosapentaenoic acid, but not of α-linolenic acid. This suggests that the observed effects are due to the VLC n-3 PUFA rather than to α-linolenic acid per se. Thus, the likely explanation for the lack of anti-inflammatory effect of α-linolenic acid at modest and even at rather high intakes is that there has been insufficient conversion to the more active EPA.
Mechanisms of action
It is clear that one anti-inflammatory mechanism of action of VLC n-3 PUFA is antagonism of production of inflammatory eicosanoids from arachidonic acid coupled with the generation of less potent EPA-derived eicosanoids and, in some conditions, anti-inflammatory resolvins(Reference Arita, Yoshida and Hong225). Altered eicosanoid profiles may have downstream effects since some eicosanoids regulate production of inflammatory cytokines. Thus, an n-3 PUFA-induced decrease in eicosanoid production might affect production of TNF-α, IL-1β, IL-6, etc. However, the effects of n-3 PUFA on inflammatory cytokine production and on some other inflammatory processes appear to be eicosanoid independent. One alternative candidate mechanism of action is altered activation of transcription factors involved in inducing transcription of inflammatory genes (e.g. NF-κB, PPAR-γ; see Calder(Reference Calder226, Reference Calder227)), which may occur as a result of altered plasma membrane signalling processes(Reference Lee, Plakidas and Lee228–Reference Weatherill, Lee and Zhao231).
Effects on clinical outcomes in inflammatory conditions
Inflammatory bowel diseases
Dietary fish oil shows improvements in animal models of IBD (see Calder(Reference Calder173)). Shoda et al. (Reference Shoda, Matsueda and Yamato232) determined that the increased incidence of CD in Japan was significantly associated with an increase in the ratio of n-6 to n-3 PUFA in the diet. They suggest that a diet high in n-6 PUFA relative to n-3 PUFA somehow plays a causal role in the disease, and that an increase in n-3 PUFA intake may be of benefit. VLC n-3 PUFA are incorporated into gut mucosal tissue of patients with IBD, who supplement their diet with fish oil and there are reports that this results in anti-inflammatory effects, such as decreased LTB4 production by neutrophils and colonic mucosa, decreased PGE2 and TXB2 production by colonic mucosa and decreased production of PGE2 by blood mononuclear cells (see Calder(Reference Calder173) for references). Small open-label or pilot studies reported clinical benefit of fish oil supplementation in UC (see Calder(Reference Calder173)). A number of randomised, placebo-controlled, double-blind studies of fish oil in IBD have been reported (see Calder(Reference Calder173) for details). The dose of VLC n-3 PUFA used in these trials was between 2·7 and 5·6 g/d and averaged about 4·5 g/d. Some of these trials indicate benefits of fish oil, which include improved clinical score, improved gut mucosal histology, improved sigmoidoscopic score, lower rate of relapse and decreased use of corticosteroids. One study of special note is that of Belluzzi et al. (Reference Belluzzi, Brignola and Campieri233) in which patients with CD in remission were randomised to receive placebo or 2·7 g VLC n-3 PUFA/d from an enterically coated fish oil preparation for 1 year: there was a significant difference in the proportion of patients who relapsed over 12 months: 28 % in the fish oil group v. 69 % in the placebo group. Reviews of trials of fish oil in IBD conclude that there is some benefit from fish oil in IBD. A recent meta-analysis has concluded that there may be reduction in requirement for corticosteroids(Reference MacLean, Mojica and Morton234).
Asthma
There are epidemiologic data linking high n-6 PUFA or low n-3 PUFA consumption with childhood asthma and allergic conditions (see Calder(Reference Calder173) for references). Early exposure to VLC n-3 PUFA does appear to alter cytokine production by neonatal T-cells(Reference Dunstan, Mori and Barden235, Reference Dunstan, Mori and Barden236) although the longer-term clinical impact of this is not yet clear. Several studies report anti-inflammatory effects of fish oil in patients with asthma, such as decreased four-series LT production and leucocyte chemotaxis (see Calder(Reference Calder173) for references). A number of randomised, placebo-controlled, double-blind studies of fish oil in asthma have been reported (see Calder(Reference Calder173) for details). A systematic review concluded that there was no consistent effect on forced expiratory volume at one second, peak flow rate, asthma symptoms, asthma medication use or bronchia hyper-reactivity(Reference Thien, Woods and De Luca237). However, one study in children has shown improved peak flow and reduced asthma medication use with fish oil(Reference Nagakura, Matsuda and Shichijyo238). A more recent report covering twenty-six studies (both randomised, placebo-controlled and others) has concluded that no definitive conclusion can yet be drawn regarding the efficacy of VLC n-3 fatty acid supplementation as a treatment for asthma in children and adults(Reference Schachter, Reisman and Tran239). However, studies by Broughton et al. (Reference Broughton, Johnson and Pace240) and Nagakura et al. (Reference Nagakura, Matsuda and Shichijyo238) indicate that there may be subgroups of asthmatic subjects who may benefit greatly from VLC n-3 PUFA. A recent study has reported the effects of a high level of fish oil, providing 5·2 g/d EPA+DHA, on sputum inflammatory cytokines and pulmonary function in exercise-induced bronchoconstriction(Reference Mickleborough and Rundell68). Concentrations of TNF-α and IL-1β in sputum supernatant were significantly lower in the fish oil group and this change was accompanied with improved pulmonary function.
Chronic obstructive pulmonary disease
Two recent studies have investigated the effects of fairly low supplemental intake of EPA+DHA on inflammatory markers, as well as exercise capacity, in COPD(Reference Broekhuizen, Wouters and Creutzberg241, Reference Matsuyama, Mitsuyama and Watanabe242). Broekhuizen et al. (Reference Broekhuizen, Wouters and Creutzberg241) reported improved exercise capacity in a cycling test but no effect on systemic levels of IL-6 and TNF-α following intake of a supplement containing 1·0 g/d EPA+DHA. Matsuyama et al. (Reference Matsuyama, Mitsuyama and Watanabe242) measured both systemic (serum) and sputum TNF-α and IL-8 levels in a 2-year intervention with a supplemental intake of 0·6 g/d EPA+DHA. The present study also failed to find differences in serum cytokine concentrations, but did report decreased sputum TNF-α and IL-8 levels in the n-3 PUFA group. This effect was accompanied by improved exercise capacity in a walk test.
Rheumatoid arthritis
Dietary fish oil shows improvements in animal models of RA (see Calder(Reference Calder173)). Several studies report anti-inflammatory effects of fish oil in patients with RA, such as decreased LTB4 production by neutrophils and monocytes, decreased IL-1 production by monocytes, decreased plasma IL-1β concentrations, decreased serum CRP concentrations and normalisation of the neutrophil chemotactic response (see Calder(Reference Calder173) for references). A number of randomised, placebo-controlled, double-blind studies of fish oil in RA have been reported (see Calder(Reference Calder173) for details). The dose of VLC n-3 PUFA used in these trials was between 1·6 and 7·1 g/d and averaged about 3·5 g/d. Almost all of these trials showed some benefit of fish oil, including reduced duration of morning stiffness, reduced number of tender or swollen joints, reduced joint pain, reduced time to fatigue, increased grip strength and decreased use of non-steroidal anti-inflammatory drugs. One study reported greater efficacy of fish oil against a background diet designed to be low in arachidonic acid(Reference Adam, Beringer and Kless243). Reviews of the trials of fish oil in RA have concluded that there is benefit and a meta-analysis concluded that dietary fish oil supplementation significantly reduces tender joint count and morning stiffness(Reference Fortin, Lew and Liang244). A recent meta-analysis has concluded that VLC n-3 fatty acids may reduce requirements for corticosteroids(Reference MacLean, Mojica and Morton234). Thus, there is reasonably strong evidence that VLC n-3 PUFA have some clinical benefits in RA.
Psoriasis
Intravenous fish oil led to resolution of psoriasis in one study(Reference Mayser, Mrowietz and Arenberger245). Dietary supplementation studies with fish oil do not present a clear picture although some of these have reported clinical benefit (see Ziboh(Reference Ziboh and Kremer246) for a review).
UV-induced skin inflammation
In experimental studies of skin inflammation, UV-induced dose–response curves were generated before and after 3 months of dietary supplementation with fish oil(Reference Rhodes, O'Farrell and Jackson247). There was a significant inhibition of the erythemal response, reduction in PGE2 release and biochemical analysis of skin biopsies, which showed that the VLC n-3 PUFA were acting as a scavenger for reactive oxygen(Reference Rhodes, O'Farrell and Jackson247, Reference Rhodes, Durham and Fraser248).
CVD
Substantial evidence from epidemiological and case-control studies indicates that consumption of fish, fatty fish and VLC n-3 PUFA reduces risk of cardiovascular mortality(Reference Bucher, Hengstler and Schindler177, Reference Studer, Briel and Leimenstoll178, Reference Kris-Etherton, Harris and Appel249, Reference Calder250). Secondary prevention studies using VLC n-3 PUFA in myocardial infarction survivors have shown a reduction in total mortality and cardiovascular mortality with an especially potent effect on sudden death(175). This protective effect of n-3 PUFA towards sudden death was apparent within 4 months of beginning supplementation(Reference Marchioli, Barzi and Bomba176). VLC n-3 PUFA have been shown to influence a number of CVD risk factors (see Kris-Etherton et al. (Reference Kris-Etherton, Harris and Appel249) and Calder(Reference Calder250) for references), and the extent to which a reduction in inflammation protects against the growth of the atherosclerotic plaque and decreases the risk and severity of cardiovascular events is not clear. However, one study suggests that VLC n-3 PUFA might act to stabilise advanced atherosclerotic plaques, perhaps through their anti-inflammatory effects(Reference Thies, Garry and Yaqoob174).
Antioxidant vitamins (C and E, β-carotene)
Oxidative stress and inflammation
There is a strong interaction between oxidative stress and inflammation. The generation of oxidants (e.g. superoxide radicals, hydrogen peroxide), for example by granulocytes exposed to bacterial cell wall peptides or lipopolysaccharides, is part of the host inflammatory response. Oxidants can damage components of host cells, such as the PUFA in cell membranes. In turn, oxidants and oxidised cell components, acting through transcription factors such as NF-κB, induce production of inflammatory eicosanoids and cytokines (Fig. 4). Thus, one mechanism to diminish inflammatory mediator production may be to prevent oxidative stress. This is accomplished through enhancing antioxidant defence mechanisms. In order to monitor antioxidant defence, individual components of that defence can be measured (e.g. vitamin C, vitamin E, antioxidant enzyme activities). Additionally, the effectiveness of the non-enzymatic endogenous antioxidant network can be assessed measuring total antioxidant capacity (TAC), defined as the moles of oxidants neutralised by 1 litre of the tested sample(Reference Serafini and Del251). TAC considers the cumulative action of all the antioxidants present in the matrix (plasma, saliva, diet, etc.), thus providing an integrated parameter rather than the simple summary of measurable antioxidants, giving an insight into the delicate balance between antioxidants molecules. Endogenous TAC has been shown to be modulated by dietary ingestion of plant food(Reference Serafini, Bugianesi and Maiani252) and to decline in subjects affected by CVD(Reference Vassalle, Petrozzi and Botto253) and cancer(Reference Ching, Ingram and Hahnel254). Population-based case-control studies have shown an inverse association of the TAC of the diet with risk of gastric cancer(Reference Serafini, Bellocco and Wolk255) and overall mortality(Reference Agudo, Cabrera and Amiano256). Despite these promising results, evidence about the role of the dietary or endogenous TAC and inflammation is lacking. Recently, TAC has been shown to be inversely and independently associated with plasma levels of CRP in 243 non-diabetic subjects(Reference Brighenti, Valtuena and Pellegrini257). This observation highlights the interrelationship between oxidative stress and inflammation and suggests that improving TAC by dietary means may be a fruitful anti-inflammatory approach.
Vitamin C
Description, sources, normal intakes and roles
The term vitamin C includes ascorbic acid (AA) and dehydroascorbic acid, both of which exhibit anti-scorbutic activity. AA, the enolic form of an α-ketolactone, is the functional and primary in vivo form of vitamin C. AA contains an asymmetric carbon atom that allows two enantiomeric forms, of which the l-form is naturally occurring. The two enolic hydrogen atoms of AA provide the electrons for its function as a reductant and antioxidant. The one-electron oxidation product of AA is the ascorbyl radical. It readily dismutates to AA and dehydroascorbic acid, the two-electron oxidation product. Both ascorbyl radical and dehydroascorbic acid are readily reduced back to AA. Dehydroascorbic acid can also be irreversibly hydrolysed to 2,3-diketo-l-gulonic acid, which is further decarboxylated and degraded to oxalic acid and threonic acid.
Vitamin C is essential for human subjects and a few other mammals, including guinea pigs and monkeys, because they are unable to synthesise AA from glucose due to lack of the enzyme gulonolactone oxidase. The main dietary sources of vitamin C in the typical Western diet are fruits and vegetables, including fruit and vegetable juices, as well as potatoes. Black currant, raw pepper, raw broccoli, kiwi, strawberries, oranges and freshly squeezed orange juice are particularly rich in vitamin C.
Present dietary reference intakes have been set at 90 and 75 mg/d for adult males and females, respectively(258). An additional 35 mg/d is recommended for smokers, given the greater metabolic turnover of vitamin C in smokers. The recommended five servings per day of fruits and vegetables should provide at least 200 mg of vitamin C. Storage and food processing have a major impact on vitamin C content. There are also an increasing number of foods and drinks fortified with vitamin C. At usual dietary vitamin C intakes, in the range of 30–180 mg/d, approximately 70–90 % is absorbed, while absorption decreases to about ≤ 50 % at doses exceeding 1 g(Reference Levine, Conry-Cantilena and Wang259).
Maximal neutrophil concentrations with minimal urinary losses are thought to be the best biomarkers for estimating vitamin C requirements. Saturation of leucocytes with vitamin C occurs at a lower intake compared with plasma(Reference Levine, Conry-Cantilena and Wang259). Mean plasma concentrations of healthy subjects are about 65 μmol/l, while scurvy occurs at plasma concentrations of less than 10 μmol/l.
Vitamin C plays an important role in mechanisms involved in immune function and inflammatory processes, including, for instance, free radical scavenging and protection against lipid peroxidation. Given its high reducing power, vitamin C is a potent water-soluble antioxidant that acts in the aqueous phase both intra- and extracellularly. It readily scavenges ROS and reactive nitrogen species including hydroxyl, peroxyl, superoxide, nitroxide radical, peroxynitrite, as well as singlet oxygen and hypochlorite(Reference Frei, England and Ames260). Ascorbyl radical, the one-electron oxidation product, and dehydroascorbic acid, the two-electron oxidation product of vitamin C, are readily regenerated to AA by glutathione and NADH or NADPH.
AA tissue levels are relatively high and thus ensure efficient antioxidant protection, for instance, against ROS produced in activated neutrophils during phagocytosis(Reference Levine, Dhariwal, Wang and Frei261). AA also scavenges ROS in the aqueous phase before they initiate lipid peroxidation of fatty acids in plasma and LDL(Reference Frei, Stocker and Ames262, Reference Jialal, Vega and Grundy263). Moreover, AA enhances antioxidant protection by sparing or regenerating α-tocopherol and glutathione back to their active forms(Reference Halpner, Handelman and Belmont264).
In addition to its antioxidant action, vitamin C is a cofactor for enzymes involved in the biosynthesis of collagen, carnitine and neurotransmitters as well as corticosteroids, the microsomal drug-metabolising enzymes and cytochrome P-450 electron transport. It modulates iron absorption, transport and storage. AA has also been shown to modulate PG synthesis(Reference Horrobin265).
Effects on inflammatory processes and mechanisms of action
In infectious diseases and inflammation, ROS are generated during neutrophil activation and phagocytosis. AA exerts cellular protection against ROS generated by the respiratory burst. It neutralises hypochlorous acid, a powerful ROS generated by myeloperoxidase in activated neutrophils and monocytes such that inactivation of α1-proteinase inhibitor by hypochlorous acid is inhibited and, consequently, proteolytic damage is prevented at the site of inflammation(Reference Halliwell, Wasil and Grootveld266). It is important to note that, under normal conditions, phagocyte-derived ROS are neutralised without inhibiting the bactericidal activity of the phagosome(Reference Anderson and Lukey267). Vitamin C also affects antimicrobial and natural killer cell activities, lymphocyte proliferation, chemotaxis and delayed-type hypersensitivity. Vitamin C further enhances phagocytosis and migration of neutrophils(Reference Goetzl, Wasserman and Gigli268). In healthy subjects, vitamin C concentrations are inversely correlated with CRP and 8-epiPGF2α(Reference Sanchez-Moreno, Cano and de Ancos269, Reference Sanchez-Moreno, Dashe and Scott270). Plasma 8-epiPGF2α and MCP-1 concentrations decreased significantly during consumption of approximately 72 mg/d vitamin C from vegetable soup(Reference Sanchez-Moreno, Cano and de Ancos269).
An energy-dependent transport system allows for a 25- to 80-fold accumulation of AA in neutrophils, lymphocytes and mast cells as compared with plasma levels(Reference Levine, Conry-Cantilena and Wang259, Reference Evans, Currie and Campbell271). During the course of infections, there is a rapid depletion of cellular ascorbate that returns to normal after recovery. At the site of inflammation, the ratio of oxidised to reduced ascorbate (dehydroascorbic acid: AA) is increased, as has been shown for the synovial fluid of RA joints(Reference Lunec and Blake272), and in the epithelial lining fluid of the lung and in the plasma of patients with adult respiratory distress syndrome(Reference Cross, Forte and Stocker273). Dehydroascorbic acid is the form of vitamin C that crosses the cell membranes and is then reduced intracellularly to AA. Reduction in vitamin C is associated with oxidation of reduced glutathione (GSH) to its oxidised form (GSSG).
Effects in inflammatory conditions
Inflammatory bowel diseases
Serum and leucocyte ascorbate levels are low in adult and pediatric patients with CD, both in active disease and in disease remission(Reference Imes, Dinwoodie and Walker274–Reference Wendland, Aghdassi and Tam277). Colonic biopsies of inflamed v. non-inflamed areas in patients with CD showed a 35 % reduction in reduced and total AA content and in patients with UC total AA content was reduced by 73 % and reduced AA by 41 %(Reference Buffinton and Doe278). Also, different biomarkers of lipid peroxidation are higher(Reference Wendland, Aghdassi and Tam277) and decreased during supplementation of vitamin C and E(Reference Aghdassi, Wendland and Steinhart279). Fish oil plus antioxidant treatment (90 mg vitamin C, 30 mg vitamin E, 150 μg retinol equivalents as β-carotene) of patients with CD showed a significant reduction in the production of PGE2 and IFN-γ by circulating monocytes and macrophages(Reference Trebble, Arden and Wootton280).
Lung inflammation
Evidence of AA oxidation has been seen in adult respiratory distress syndrome(Reference Cross, Forte and Stocker273). Adults with mild asthma exhibit decreased AA in lung fluid and increased GSSG concentrations which, in the presence of normal plasma concentrations, are suggestive of oxidative stress in the airways(Reference Kelly, Mudway and Blomberg281). Cross-sectional studies showed an inverse relation between plasma vitamin C and lung inflammation(Reference Winklhofer-Roob, Ellemunter and Fruhwirth282) and suggest that high plasma AA concentrations or vitamin C intakes have a positive impact on lung function(Reference Schwartz and Weiss283, Reference Britton, Pavord and Richards284). Compared with the highest quintile, serum vitamin C concentrations in the lowest quintile were associated with an increased risk of asthma in children and adolescents(Reference Harik-Khan, Muller and Wise285). Low maternal vitamin C intake during pregnancy is associated with asthma in 5-year-old children(Reference Devereux, Turner and Craig286). Asthmatic children showed reduced vitamin C concentrations in leucocytes (50 %) and plasma (35 %)(Reference Aderele, Ette and Oduwole287). Small clinical studies showed that vitamin C supplements of 2 g/d exert protective effects on airway responsiveness to viral infections and allergens(Reference Bucca, Rolla and Farina288), but have little effect on exercise-induced asthma(Reference Cohen, Neuman and Nahum289), although one study did report attenuated exercise-induced bronchoconstriction in patients with asthma(Reference Tecklenburg, Mickleborough and Fly290).
Rheumatoid arthritis
Evidence of AA oxidation has been seen in RA(Reference Lunec and Blake272). An intervention study with a Mediterranean diet, rich in antioxidant containing foods, in RA showed a negative correlation between plasma vitamin C levels and disease activity(Reference Hagfors, Leanderson and Skoldstam291).
CVD
Vitamin C and other single vitamin supplementation and CVD have been reviewed recently(Reference Traber292). Associations between antioxidants and markers of endothelial function (sICAM-1; flow-mediated vasodilation) and inflammation (CRP, fibrinogen and white blood corpuscles count) were studied in subjects recruited from the general population. Plasma vitamin C and CRP concentrations were inversely related(Reference van Herpen-Broekmans, Klopping-Ketelaars and Bots293). Recent randomised controlled trials have shown that vitamins C plus E improved arterial stiffness and endothelial function in patients with essential hypertension(Reference Plantinga, Ghiadoni and Magagna294), vitamin C decreased sPselectin concentration and vitamins C plus E decreased sICAM-1 in patients with chronic degenerative aortic stenosis with or without concomitant coronary artery disease(Reference Tahir, Foley and Pate295).
Vitamin E
Description, sources, normal intakes and roles
Vitamin E is an umbrella term for a number of tocopherols and tocotrienols. They are composed of a chromanol ring and a 16-C phytyl tail, which is saturated in the tocopherols and unsaturated in the tocotrienols. The chromanol ring is substituted with one, two or three methyl groups and one hydroxyl group, which exerts the antioxidant function. Depending on the number and positions of the methyl groups of the chromanol ring, α- (three methyl groups at C-5, C-7 and C-8 positions), β- (two methyl groups at C-5 and C-8 positions), γ- (two methyl groups, C-5 position unsubstituted) and δ- (one methyl group at C-8 position) forms can be distinguished for both tocopherols and tocotrienols. These differences in the chemical structure have an impact not only on the hydrophobicity of the different types of tocopherols, but also on the discrimination by the α-tocopherol-transfer protein in the liver, which recognises the fully methylated aromatic ring of α-tocopherol(Reference Traber, Burton and Ingold296). Consequently, α-tocopherol is incorporated into nascent VLDL, while γ-tocopherol is metabolised and excreted, resulting in substantially lower γ- than α-tocopherol concentrations in plasma and tissues. The physiological metabolites of tocopherols and tocotrienols, i.e. the carboxyethyl hydroxychromans (CEHC), are generated via a similar degradation pathway that involves cytochrome P450-mediated n-oxidation of the phytyl chain and subsequent β-chain oxidation to the corresponding hydrophilic metabolites in the absence of modification of the chromanol ring(Reference Schultz, Leist and Elsner297, Reference Lodge, Ridlington and Leonard298). The hydroxyl group on the chromanol ring exerts the antioxidant action. It can be esterified to give rise to tocopherol acetate or succinate.
Vitamin E is widely distributed in different kinds of foods. Dietary vitamin E mainly consists of α- and γ-tocopherol. Frequently, the major source of vitamin E is γ-tocopherol, which is present at high concentrations in plant seeds and vegetable oils such as maize oil and soyabean oil, as well as in nuts such as walnuts, pecans and peanuts(Reference McLaughlin and Weihrauch299, Reference Lehmann, Martin and Lashley300). Fats and oils contributed about 20 %, vegetables 15 %, meat and fish 12·5 %, desserts 10 %, breakfast cereals 9 %, fruits 5 %, bread and grain products 5 %, dairy products 4·5 %, mixed main dishes 4 %, nuts and seeds 4 %, eggs 3 %, salty snacks 3 %, legumes 2 % and soups, sauces and gravies 2 % of the total vitamin E intake among American adults(Reference Murphy, Subar and Block301). North American dietary reference intakes for vitamin E have recently been set at 15 mg/d for both men and women(258). Vitamin E supplements further contribute to vitamin E intake. In the Boston Nutritional Status Survey, 38 % of the men of 60 years of age and older and 49 % of the women were users, and 68 % and 73 %, respectively, of the male and female users took vitamin E supplements(Reference Slesinski, Subar and Kahle302). In contrast to vitamin E in the diet, almost all supplements contain α-tocopherol exclusively.
Vitamin E is a potent chain-breaking antioxidant that acts mainly in the lipid phase and interrupts the chain reaction of lipid peroxidation and, consequently, prevents the propagation of free radical-initiated reactions(Reference Burton and Ingold303, Reference Kamal-Eldin and Appelqvist304). There are differences between α- and γ-tocopherols(Reference Kamal-Eldin and Appelqvist304) and a recent review has paid attention to γ-tocopherol(Reference Jiang, Christen and Shigenaga305). α-Tocopherol decreases lipid peroxidation and enhances LDL resistance to oxidation(Reference Winklhofer-Roob, Rock and Ribalta306, Reference Winklhofer-Roob, Ziouzenkova and Puhl307). γ-Tocopherol decreases the concentrations of biomarkers of oxidative stress, including LDL oxidation, MDA formation and superoxide anion generation and is more potent than α-tocopherol(Reference Saldeen, Li and Mehta308, Reference Liu, Wallin and Wallmon309). α-Tocopherol also decreased the expression of the scavenger receptors SR-A and CD36, which are important in the formation of foam cells in the early stages of atherogenesis(Reference Devaraj, Hugou and Jialal310). However, γ-tocopherol cannot replace α-tocopherol in its functions in fertility(Reference Bieri and Evarts311).
A more recently discovered mechanism of action has been restricted to γ-tocopherol and its physiological metabolite, γ-CEHC. Because of an unsubstituted position on the chromanol ring, γ-tocopherol traps certain reactive nitrogen species in cell lines and unsaturated liposomes with formation of 5-nitro-γ-tocopherol(Reference Cooney, Franke and Harwood312, Reference Christen, Woodall and Shigenaga313).
Effects on inflammatory processes and mechanisms of action
One of the mechanisms of action of vitamin E on inflammatory processes is its well-known antioxidant activity, which limits the generation of ROS as well as the perpetuation and damaging effects of the reactions initiated by ROS, which in turn, could activate the inflammatory process.
α-Tocopherol, especially at high doses, decreases the release of pro-inflammatory cytokines, the chemokine IL-8 and PAI-1 and decreases adhesion of monocytes to endothelium(Reference Devaraj and Jialal314). Vitamin E (particularly γ-tocopherol) has been shown to decrease NF-κB activation(Reference Sundl, Gruber and Roob315). Additional effects include inhibition of the activities of protein kinase C, 5-LOX, tyrosine kinase and COX-2(Reference Tasinato, Boscoboinik and Bartoli316, Reference Devaraj and Jialal317). α-Tocopherol has also been shown to decrease CRP levels(Reference Devaraj and Jialal314).
γ-Tocopherol and γ-CEHC exert actions not shared by α-tocopherol and α-CEHC. For instance, γ-tocopherol but not α-tocopherol inhibits PGE2 generation in cell culture(Reference Jiang, Lykkesfeldt and Shigenaga318–Reference Jiang and Ames320). Furthermore, LTB4, a potent chemotactic agent synthesised by 5-LOX of neutrophils, and TNF-α were decreased in carrageenan-induced inflammation in rats and in different cell lines by γ-tocopherol, but not by α-tocopherol(Reference Grammas, Hamdheydari and Benaksas319, Reference Jiang and Ames320). γ-Tocopherol decreased oxidised LDL-mediated NF-κB acitivation in human coronary artery endothelial cells(Reference Li, Saldeen and Mehta321). γ-Tocopherol and γ-CEHC, but not α-tocopherol, inhibited COX activity in macrophages and epithelial cells(Reference Jiang, Elson-Schwab and Courtemanche322).
Effects in inflammatory conditions
Rheumatoid arthritis
Low antioxidant level (the lowest v. higher tertiles of the product of molar concentrations of α-tocopherol, β-carotene and selenium) was a risk factor for RA over 20 years of follow-up of a case-control study(Reference Heliovaara, Knekt and Aho323). In the KRN/NOD mouse, a mouse model of RA, vitamin E (RRR α-tocopherol) prevented joint destruction as assessed by histological analysis, but did not have an effect on clinical features such as time of onset or disease intensity. In addition, levels of IL-1β, the main cytokine involved in articular destruction, were reduced, as was neutrophil activation measured by chemiluminescence(Reference De Bandt, Grossin and Driss324). In an animal model for RA, mice fed a fish oil and additional vitamin E-enriched diet showed significantly lower serum IL-6, TNF-α, PGE2, TXB2 and LTB4 levels compared with the fish oil diet alone(Reference Venkatraman and Chu325). In a double-blind placebo-controlled trial patients with RA, who received 600 mg vitamin E twice a day, showed a significant pain reduction but no changes in clinical and biochemical indices of inflammation(Reference Edmonds, Winyard and Guo326).
CVD. Vitamin E inhibits LDL oxidation both when induced by copper ions ex vivo (Reference Winklhofer-Roob, Ziouzenkova and Puhl307, Reference Dieber-Rotheneder, Puhl and Waeg327) and in cell culture(Reference Steinbrecher, Parthasarathy and Leake328). In addition to that vitamin E could have an effect on different aspects of atherogenesis, given that it (i) inhibits smooth muscle cell proliferation by inhibiting protein kinase C(Reference Azzi, Boscoboinik and Marilley329); (ii) inhibits platelet adhesion, aggregation and release(Reference Freedman, Farhat and Loscalzo330); (iii) decreases monocyte adhesion to the endothelium by downregulating the expression of adhesion molecules and reducing monocyte superoxide production(Reference Devaraj, Li and Jialal331); (iv) increases prostacyclin synthesis in human endothelial cells, which is a potent vasodilator and inhibitor of platelet aggregation(Reference Chan and Leith332, Reference Tran and Chan333); (v) mediates upregulation of the expression of cytosolic phospholipase A2 and COX(Reference Chan, Wagner and Kennedy334); and (vi) inhibits the expression of ICAM-1 and VCAM-1 in endothelial cells in culture(Reference Cominacini, Garbin and Pasini335).
While epidemiological studies consistently showed an inverse correlation between vitamin E status and risk of CVD, the results of vitamin E supplementation trials have been much less convincing regarding prevention of CVD. A possible explanation could be the fact that only α-tocopherol, which is almost exclusively present in vitamin E supplements, has been taken in these studies. This is further supported by the observation that γ-, not α-tocopherol serum levels are reduced in CHD(Reference Ohrvall, Sundlof and Vessby336). It is known from several intervention trials(Reference Handelman, Machlin and Fitch337, Reference Sundl, Resch and Bergmann338) that α-tocopherol supplementation causes a marked drop in γ-tocopherol concentrations in plasma and lipoproteins.
Carotenoids
Description, sources, normal intakes and roles
Carotenoids represent a group of over 600 coloured pigments found in nature that are responsible for the typical colour of fruits and vegetables as well as some animals. Carotenoids are composed of a 40-carbon atom skeleton and are either hydrocarbons (also called xanthophylls) that contain only H and C (α-, β-carotene, lycopene), or oycarotenoids that contain also O (lutein, zeaxanthin). Provitamin A carotenoids (α- and β-carotene, and β-cryptoxanthin) are carotenoids that can be cleaved in the intestine either by central cleavage, giving rise to two molecules of retinol from one molecule β-carotene, or excentric cleavage, giving rise to one molecule of retinol and carotenoid cleavage products.
Plasma carotenoid concentrations are useful biomarkers of the consumption of fruits and vegetables(Reference Drewnowski, Rock and Henderson339). The most prevalent carotenoids in the Western diet are α-carotene, β-carotene, lycopene, β-cryptoxanthin, lutein and zeaxanthin. Dietary sources of α-carotene are carrots, sweet potatoes, cabbage, spinach, pumpkin, broccoli and pepper; of β-carotene are carrots, pumpkin, red pepper, green beans, green salad, broccoli and melon; of β-cryptoxanthin are avocado, green olives, peach, mandarin, mango, papaya and orange; of lycopene are tomatoes and tomato products, and guave; of lutein are dark green leafy vegetables such as cabbage, spinach, broccoli, peas, cauliflower and green salad; of zeaxanthin is maize. Most of the naturally occuring carotenoids are present as all-trans isomers and are transformed into the cis isomers during food processing (heating; lycopene) or in the intestinal mucosa cells (β-carotene). Astaxanthin is found in marine animals and vegetables.
As suggested by Krinsky(Reference Krinsky340), the biological activities of carotenoids should be diffentiated as:
(i) functions, including acting as accessory pigments in photosynthesis, protection against photosensitisation and role as provitamin A;
(ii) actions, including antioxidant, immunoenhancing, anti-mutagenic and anti-carcinogenic actions;
(iii) associations, including decreasing risk of macular degeneration and cataracts, some cancers and some cardiovascular events.
Even though carotenoids are recognised mainly for their provitamin A functions, less than 10 % of them exert provitamin A activities in mammals. Of the carotenoids, β-carotene has the highest provitamin A activity. The efficiency of β-carotene conversion to vitamin A decreases with higher β-carotene intake. Lutein is among the most prevalent carotenoids found in normal skin and a major carotenoid in the macular pigment and effectively filters blue light and thus protects the retina from light-induced oxidative damage(Reference Krinsky341).
Effects on inflammatory processes and mechanisms of action
Carotenoids possess immunomodulatory activities in human subjects and animals, including, for instance, stimulation of the phagocytic and bacteria-killing ability of peripheral blood neutrophils and peritoneal macrophages and of lymphocyte blastogenesis, increasing the population of specific lymphocyte subsets and lymphocyte cytotoxic activity, as well as stimulation of the production of various cytokines. β-Carotene inhibits inflammatory gene expression by suppressing the activation of the redox-sensitive transcription factor, NF-κB(Reference Bai, Lee and Na342, Reference Flohé, Brigelius-Flohé and Saliou343).
Effects in inflammatory conditions
While β-carotene has been studied quite extensively, data are limited for other carotenoids. In Table 2, the effects of carotenoids as well as of vitamins C and E are listed. Biomarkers of inflammation have been included in a variety of studies and quite consistently either showed an inverse relation between status of different carotenoids and inflammation or reduction in elevated biomarkers in response to intervention using carotenoid-rich foods or supplements. Clinical end point studies are particularly scarce. Like other serum antioxidants such as vitamin C, plasma carotenoid concentrations decline during the acute-phase response to infection and injury in the presence of increased levels of biomarkers of inflammation(Reference Louw, Werbeck and Louw344). Inverse relations between plasma β-carotene concentrations and biomarkers of inflammation have been shown in a re-evaluation study of the National Health and Nutrition Examination Survey III study(Reference Kritchevsky, Bush and Pahor345), as well as in studies in critically ill patients(Reference Quasim, McMillan and Talwar346). The underlying mechanisms are not known, but may involve a cellular carotenoid-binding protein(Reference Lakshman and Rao347). Most importantly, however, β-carotene and retinol levels are restored after resolution of the inflammatory state in the absence of β-carotene and/or retinol supplements. Study of the effects of lycopene on murine bone marrow-derived DC, which are the most potent antigen-presenting cells, revealed that lycopene downregulates NF-κB as well as mitogen-activated kinases such as extracellular signal-regulated kinase 1/2, p38 and jun N-terminal kinase(Reference Kim, Kim and Ahn348). Astaxanthin showed a dose-dependent anti-inflammatory effect in endotoxin-induced uveitis in rats by suppressing NO, PGE2 and TNF-α production(Reference Ohgami, Shiratori and Kotake349). Mice fed lycopene showed significant inhibition of ear swelling induced by UV radiation(Reference Lee, Faulhaber and Hanson350). In a RCT, high intake of carotenoid-rich fruits and vegetables reduced CRP concentrations(Reference Watzl, Kulling and Möseneder351). In another RCT, dietary intake of a tomato-based drink (providing 5·7 mg lycopene, 1 mg β-carotene, 3·7 mg phytoene and 2·7 mg phytofluene and 1·8 mg α-tocopherol per day), was associated with 34 % decrease in TNF-α in whole blood(Reference Riso, Visioli and Grande352).
Table 2 Summary of studies investigating the anti-inflammatory actions of vitamins C and E and of carotenoids

LT, leukotriene; COX, cyclo-oxygenase; CEHC, carboxyethyl hydroxychroman; ERK, extracellular signal-regulated kinase; JNK, jun N-terminal kinase; iNOS, inducible NO synthase; RA, rheumatoid arthritis; ICAM, intercellular adhesion molecule; CRP, C-reactive protein; WBC, white blood cell count; NHANES, National Health and Nutrition Examination Survey; EPIC, European Prospective Investigation of Cancer; IBD, inflammatory bowel disease; IFN, interferon; MDA, malondialdehyde.
Inflammatory bowel diseases. Plasma concentrations of different carotenoids are decreased in patients with CD(Reference Wendland, Aghdassi and Tam277, Reference Rumi, Szabo and Vincze353). In a rat model of colitis, lycopene efficiently counteracted the inflammatory response and mucosal damage in iron-supplemented animals(Reference Reifen, Nur and Matas354), which can be explained by the fact that transition metal iron can give rise to the formation of hydroxyl radical, the most reactive and cytotoxic ROS that has the capacity of oxidising biomolecules including PUFA, proteins and DNA bases and that lycopene acts as an antioxidant.
Asthma. Whole blood concentrations of total carotenoids and lycopene were decreased in patients with asthma compared with controls and correlated with sputum concentrations of carotenoids(Reference Wood, Garg and Blake355).
Rheumatoid arthritis. An inverse association between serum β-cryptoxanthin levels and RA has been reported in women aged 55–69 years(Reference Cerhan, Saag and Merlino356). A nested case-control study within the European Prospective Investigation of Cancer Incidence study focused on dietary intake and occurrence of new cases of inflammatory polyarthritis and showed that subjects with dietary intake of zeaxanthin and β-cryptoxanthin in the highest tertile were at lower risk of developing inflammatory polyarthritis compared with those in the lowest tertile. The effect of β-cryptoxanthin remained significant also after adjustments for total energy and protein intakes as well as for smoking(Reference Pattison, Symmons and Lunt357). In another study, plasma β-carotene concentrations preceding diagnosis of RA by 2–15 years were lower in patients who developed RA compared with controls(Reference Comstock, Burke and Hoffman358).
CVD. Association between antioxidants and markers of endothelial function (sICAM-1 and flow-mediated vasodilation) and inflammation (CRP, fibrinogen and white blood cell count) were studied in subjects recruited from the general population. Lutein and lycopene were inversely related to sICAM-1 and β-carotene was inversely related to white blood cell count and CRP(Reference van Herpen-Broekmans, Klopping-Ketelaars and Bots293). In a nested case-control study within the Women's Health Study, CVD risk was associated with plasma lycopene concentrations such that women in the upper three quartiles of plasma lycopene concentrations had a 50 % risk reduction(Reference Sesso, Buring and Norkus359). Similar results were not observed in men participating in the Physicians' Health Study(Reference Sesso, Buring and Norkus360).
Obesity. Low carotenoid and elevated CRP concentrations are frequently observed in obesity, and are attributed to unfavourable dietary habits and low-grade inflammation(Reference Suzuki, Inoue and Hioki361, Reference Wallstrom, Wirfalt and Lahmann362). An inverse correlation between plasma carotenoid concentrations and BMI was reported in obese children and adolescents(Reference Winklhofer-Roob, Sargsyan and Maritschnegg363).
Flavonoids
Description, sources, normal intakes and roles
Polyphenols are secondary metabolites of plants involved in pigmentation, reproduction and protection against pathogens(Reference Manach, Scalbert and Morand364). Presently there are more than 8000 known polyphenolic substances sharing a common chemical structure (hydroxyl group on aromatic ring) with different constituents. Flavonoids are the most abundant polyphenols present in the human diet and represents a subclass of molecules characterised with a C6–C3–C6 backbone structure(Reference Manach, Scalbert and Morand364). Flavonoids can be divided into several classes according to different constituents such as flavanones, flavone, flavanols and flavonols. They can be found in almost all plant foods and, among the flavonols, myricetin, kaempferol and quercetin are the most representative, while catechins are the most abundant flavanols contained in tea leaves. Flavanones are mainly represented in the diet by taxifolin, naringinin and hesperitin. The main sources of flavanones are citrus fruits. Flavones, luteolin, wogonin and apigenin, are less common and identified in sweet red pepper and celery. In addition to these, other classes of flavonoids are present in the diet such as proanthocyanidins and their oligomers.
Dietary intake of flavonoids, calculated in 1976 in the US(Reference Kuhnau365), was 1 g/d with 16 % as flavonols, flavones and flavanones; 20 % as catechins; 17 % as anthocyanins and 45 % as bisflavones. However, the high variability of polyphenol intake, the lack of homogeneous database and reliable biomarkers of exposure make it difficult to provide a reliable estimate of flavonoid intake in the different countries.
Over the last years, there have been significative advances in the understanding of the absorption and metabolism of flavonoids. In order to be absorbed, flavonoid glycosides, the most abundant form in food, are hydrolysed by intestinal enzymes or colon microflora. During the process of absorption, flavonoids are conjugated in the small intestine and later in the liver to increase their hydrophilicity and facilitate their elimination from the body in urine and bile. The circulating forms of flavonoids are mainly conjugated and evidence regarding their accumulation in specific target tissues is still lacking.
Flavonoids have been suggested to have anti-inflammatory activity through several action mechanisms involving the reduction in the concentration of prostanoids and leukotrienes through the inhibition of eicosanoid generating enzymes such as phospholipase A2, COX and LOX(Reference Baumann, von Bruchhausen and Wurm366).
Anti-inflammatory actions of flavonoids
Quercetin was the first flavonoid shown to inhibit neutrophil phospholipase A2(Reference Lee, Matteliano and Middleton367) with an IC50 of 57–100 μm. Flavanones such as hesperitin and naringenin were shown to be less effective inhibitors of snake venom phospholipase A2 compared with the flavonols quercetin, kaempferol and myricetin(Reference Welton, Tobias and Fiedler-Nagy368). Biflavonoids such as amentoflavone, ginkgetin, ochnaflavone and iso-ginkgetin were shown to be good inhibitors, with an IC50 of about 10 μm for phospholipase A2 from several sources. Luteolin, 3,4-dihydroxy-flavone, galangin, apigenin and morin were the first flavonoids to be shown to inhibit COX(Reference Baumann, von Bruchhausen and Wurm366). Quercetin and kaempferol are potent inhibitors of COX from rat peritoneal macrophages(Reference Welton, Tobias and Fiedler-Nagy368). The flavones apigenin, luteolin, galangin, kaempferol and quercetin and the biflavonoids amentoflavone, broussochalcone A and kuraridin are potent COX-1 inhibitors, although with different potencies. Catechin weakly inhibited COX-2 but at very high concentration (100 μm)(Reference Noreen, Serrano and Perera369). Flavonols such as kaemferol, quercetin, morin and myricetin were found to be better LOX inhibitors than flavones and with a preferential effect on 5-LOX compared with 12-LOX. Flavanones such as naringenin were not inhibitory against 5- and 12-LOX. Endothelial NOS was inhibited weakly by quercetin at very high concentration (IC50 220 μm)(Reference Chiesi and Schwaller370) with a lack of effect on nNOS and iNOS. Several flavonoids (rutin, hepseridin, catechin and tricin) had no effect on the three forms of NOS. Using LPS- or cytokine-treated macrophages or macrophage cell lines for stimulated NO production, quercetin, apigenin and luteolin were found to inhibit NO production(Reference Kim, Murakami and Nakamura371), downregulating iNOS expression. Catechins and flavanones at concentrations below 100 μm did not affect NO production by LPS-treated RAW 264·7 cells(Reference Liang, Huang and Tsai372). Glycosylated flavonoids, such as vitexin, had little effect on NO production with no effect at a concentration of 100 μm. Flavones such as apigenin, wogonin and luteolin were the most efficient inhibitors (IC50 10–20 μm). Furthermore, studies(Reference Kim, Cheon and Kim373) showed that the inhibitory effect of apigenin, genistein and kaempferol on NO production is mediated by an effect on induction of iNOS expression. Taken together, these results suggest that some flavonoids are natural inhibitors of iNOS induction, but not inhibitors of iNOS activity. With regard to inflammatory cytokines, genistein was reported to inhibit IL-1β, IL-6 and TNF-α production by LPS-stimulated human blood monocytes(Reference Geng, Zhang and Lotz374). Genistein and silybin were shown to inhibit TNF-α production from LPS-treated RAW cells(Reference Cho, Kim and Park375). Flavonoids appear to inhibit the expression of inflammation-related enzymes/proteins partly by suppressing activation of NF-κB and AP-1, an effect potentially mediated by the inhibition of different protein kinases (e.g. mitogen-activated protein kinase; extracellular signal-regulated kinase 1/2) involved in signal transduction pathway.
Quercetin has been shown to affect iNOS expression by inhibiting p38 MAPK and TNF-α from LPS-induced RAW cells by inhibiting jun N-terminal kinase/SAPK and Ap-1 DNA binding(Reference Wadsworth, McDonald and Koop376, Reference Wadsworth and Koop377). TNF-α was also inhibited through inhibition of extracellular signal-regulated kinase 1/2 and p38 MAPK. Quercetin was shown also to affect NF-κB activation by extracellular signal-regulated kinase and p38 kinase inhibition(Reference Cho, Park and Kwon378). The effect on NF-κB was shown also for genistein, apigenin, kaempferol and epigallocatechin 3-gallate. The evidence so far shows that flavonoids are able to inhibit the expression of inflammation-related enzymes/proteins partly by suppressing activation of NF-κB and AP-1, an effect potentially mediated by the inhibition of different protein kinases involved in the signal transduction pathway.
Regarding the effect of flavonoids on the expression of inflammatory markers and processes in vivo, scientific evidence is largely lacking. Quercetin inhibited release of TNF-α from carrageenan-induced air-pouch exudates in rats(Reference Morikawa, Nonaka and Narahara379). Quercetin and rutin were found to suppress lethal endotoxic shock induced by LPS in mice(Reference Takahashi, Morikawa and Kato380). Orally administered luteolin was showed to inhibit TNF-α production in LPS-treated mice(Reference Ueda, Yamazaki and Yamazaki381).
Human studies investigating the effect of flavonoids on markers of inflammation are scarce and most of them focus on the use of flavonoid-rich foods and not on pure molecules. One month of supplementation with 30 g/d ethanol as wine or as gin in healthy men was able to decrease plasma levels of IL-1α and fibrinogen(Reference Zern, Wood and Greene383). However, expression of the several adhesion molecules on monocytes and T-lymphocytes was significantly decreased only after wine ingestion. Wine also decreased serum concentrations of sVCAM-1, sICAM-1 and CRP. In another study(Reference Zern, Wood and Greene383), supplementation with a grape polyphenol extract containing anthocyanins, quercetin, myricetin, kaempferol and resveratrol (36 g/d for 4 week) produced a significant decrease in plasma TNF-α with no effect on CRP and IL-6 concentrations in both pre- and post-menopausal women. In both studies described earlier, the lack of measurement of polyphenol levels in body fluids does not allow any conclusion about the molecules responsible for the anti-inflammatory effect to be made. In an intervention study where plasma levels of flavonoids were measured, daily black tea supplementation (900 ml) for 4 weeks significantly increased plasma catechin levels (29 %) compared with baseline values but without any changes on CRP and TAC(Reference Widlansky, Duffy and Hamburg384). Moreover, in a prospective study in non-diabetic women, the intake of flavonols and flavones was not significantly associated to plasma concentrations of CRP and IL-6 or with the development of type 2 diabetes(Reference Song, Manson and Buring385). Recently, in the attempt to investigate diet-induced postprandial oxidative and inflammatory stress, acute ingestion of 50 ml of extra virgin olive oil has been shown to decrease plasma levels of inflammatory molecules (LTB4 and TXB2) together with an increase in serum TAC(Reference Bogani, Galli and Villa386).
As overall, only about 1 % of the polyphenol that is consumed is absorbed, with large differences according to the different categories, the plasma concentration that is achieved after eating polyphenol rich food is about 1 μm(Reference Manach, Scalbert and Morand364). The low concentration of flavonoids in biological fluids might present an obstacle in exerting the putative anti-inflammatory action in vivo. Human studies involving food items need to be linked with a chemical characterisation of the flavonoid composition of the food and with the assessment of specific biomarkers of single phenolics and antioxidant activity. Moreover, information obtained on the basis of acute ingestion studies might be extremely valuable for designing long-term intervention trials. On the basis of the existing evidence, a clear conclusion cannot be drawn and further human trials are needed to elucidate the role of flavonoids as anti-inflammatory agents in vivo.
The gut microflora and inflammation
Gut microflora
The intestinal epithelium of the gastrointestinal tract and its associated microflora are vital to the protection of the body(Reference Rolfe, Mackie, White and Isaacson387, Reference Holzapfel, Goktepe, Juneja and Ahmedna388). At birth, the gastrointestinal tracts of babies are sterile but then rapidly become colonised by micro-organisms from the mother's faecal and vaginal microbiota and the immediate environment. Consumption of oxygen within the gut by the first colonising organisms, facultative anaerobic bacteria, enables fully anaerobic bacteria to then colonise the lower gut. Bifidobacteria are more predominant in breast-fed babies, whereas a more complex population (similar to that of adults) develops in formula-fed babies, which comprises clostridia, bacteroides, enterobacteria, streptococci and a lower level of bifidobacteria(Reference Edwards and Parrett389). SCFA produced by bifidobacteria help to protect the baby against infection. During the first year of life, the intestinal microbiota stabilises and gradually resembles that of an adult. Up to 1014 micro-organisms are contained in the adult human body – ten times more microbial cells than human – most of these micro-organisms are located in the gastrointestinal tract(Reference Holzapfel, Haberer and Snel390). More than 1 kg of bacteria is present in the gut and faeces typically comprise 50 % bacteria, meaning that human subjects excrete 50–100 g of bacteria each day. Micro-organisms are at their lowest numbers ( < 103 CFU/g) in the stomach due to its low pH and fast transit time but reach levels as high as 1012 CFU/g in the colon, due to its much slower transit time, less acidic pH and low oxygen levels. Faecal isolates are generally representative of the microbiota of the distal colon. At least 500 different bacterial species have been cultured from faeces (including lactobacilli and bifidobacteria), yet just forty species account for 99 % of those that have been identified. The microbial inhabitants of the intestinal microbiota are still not fully identified due to the limitations of identification techniques and individual variations. New molecular typing methods are now enabling identification of further gut-associated species.
Studies with germ-free animals have demonstrated the importance of the gut microflora for the normal development of the gastrointestinal tract and the education and stimulation of the immune system(Reference Rhee, Sethupathi and Driks391–Reference Yamanaka, Helgeland and Farstad393). In the absence of an intestinal microflora, the intestinal immune system is underdeveloped and certain functional parameters, such as macrophage phagocytic ability and Ig production, are reduced(Reference Wostmann394). The intestinal microflora may also be important in protection against atopic disease(Reference Ouwehand, Isolauri and Salminen395–Reference Rautava, Ruuskanen and Ouwehand397), as demonstrated by Sudo et al. (Reference Sudo, Sawamura and Tanaka398) who showed that oral tolerance could be restored in germ-free mice by reconstitution of the intestinal microflora with bifidobacteria at the neonatal stage. The intestinal microflora of infants with atopic disease may differ from healthy infants, with higher levels of clostridia and lower levels of bididobacteria in those with atopy(Reference Kalliomaki, Kirjavainen and Eerola396).
The major function of the colonic microflora is the fermentation of dietary substances that have passed undigested through the small intestine and mucus produced within the gut. Saccharolytic fermentation produces SCFA such as acetate, propionate, butyrate, which are essential nutrients for the colonocytes. The acids have an additional protective benefit by lowering the pH of the intestinal lumen and by their antimicrobial nature. Butyric acid (produced by bifidobacteria) is particularly important because it is the preferred energy source of the epithelial cells in the colon and influences cell proliferation. It is thought to play a prominent role in reducing the risk of colon cancer. By contrast, some of the end products of proteolytic fermentation are carcinogenic(Reference Macfarlane, Macfarlane, Fuller and Perdigon399). A healthy, or balanced, intestinal microflora is thought to be one that is predominantly saccharolytic and that comprises significant numbers of bifidobacteria and lactobacilli, such that their activities predominate over those that may be harmful (e.g. clostridia, Staphylococcus aureus, toxic E. coli, sulphate-reducers and certain Bacteroides species)(Reference Cummings, Antoine and Azpiroz10, Reference Gibson and Roberfroid400–Reference Fooks, Fuller and Gibson402). Predominance of the latter groups of bacteria may predispose the host to a number of clinical disorders while increasing susceptibility to infections by transient enteropathogens(Reference Fooks, Fuller and Gibson402).
Prebiotics
Description, sources, intakes and roles
The earliest definition of a prebiotic was ‘a non-digestible food ingredient that beneficially affects the host by selectively stimulating the growth and/or activity of one or a limited number of bacteria in the colon, and thus improves host health’(Reference Gibson and Roberfroid400). As research has progressed, this definition has been refined. There are now three accepted criteria for classifying a food ingredient as a prebiotic.
(i) It should be non-digestible and non-absorbable – it should be resistant to gastric acidity, hydrolysis by intestinal brush border/pancreatic digestive enzymes and gastrointestinal absorption. This makes ‘prebiotics’ completely available for intestinal microflora.
(ii) It should be selectively fermented by the intestinal (mainly colonic) microflora, meaning that the growth of some groups of bacteria is promoted (such as members of the genus Bifidobacterium and Lactobacillus to which most probiotics belong), others are not affected (commensals such as Bacteroides, Fusarium) and still others are suppressed (pathogenic organisms such as Clostridium perfringens group, Salmonella, coliforms, etc.). This selective interaction with intestinal fermentation also results in additional production of bacterial metabolites (mainly SCFA such as acetate, propionate and butyrate) that are absorbed in the blood of the host and that interact in various ways (e.g. cell signalling function, energy substrate for certain cell types) with the host metabolism.
(iii) It should result in improvement of health-related markers, this being a direct consequence of the selective interaction of prebiotics with intestinal fermentation.
Selective prebiotic interaction with the colonic bacterial ecosystem represents a potentially important improvement of the ‘external environment’ of the host. This observation is at the basis of prebiotic action: prebiotics selectively interact with resident intestinal microflora and thereby stimulate the development and metabolic activity of organisms that belong to bacterial genera to which the most common ‘probiotics’ also belong. Moreover, while being fermented, prebiotics themselves are converted into biologically active molecules that pass into the systemic environment. These metabolites may be responsible for at least part of nutritional benefits observed upon eating prebiotics.
Inulin-type fructans are β(2-1)-linked fructose oligomers with chain length ranging between three and hundred depending on the plant source(Reference van Loo, Gibson and Rastall403). These complex carbohydrates are present in most plants that occur in regions with a moderately cold climate, characterised by periods of frost. As a consequence, they are present not only in various foodplants such as wheat, onions, garlic, chicory root, Jerusalem artichoke, etc., but also in fruits such as banana and tomato(Reference van Loo, Coussement and de Leenheer404). It has been estimated that via the consumption of food plants, people consume between 1 and 4 g of inulin per day. At present, inulin (with average chain length of ten) and its partial hydrolysate oligofructose (average chain length of four) are produced on a commercial scale via extraction from the chicory root. The latter is also enzymatically synthesised from sucrose. These products are used globally and are applied in thousands of prepared food products such as dairy, meat products, bakery, drinks, spreads, etc.
Galacto-oligosaccharides are enzymatically synthesised from lactose mainly with transferase enzymes. The link between the galactose moieties depends on the enzyme system used. Galacto-oligosaccharides formed this way are always short-chain molecules, with an average chain length of three. They do not occur as such in nature, although more complex but structurally related forms are present in mammalian breast milk. Combined with long-chain inulin fractions in milk formulations, they were shown to shift the composition of the intestinal microflora of babies close to that seen in breast-fed babies, and at the same time to induce some immunological and anti-inflammatory effects(Reference Rastall, Gibson and Rastall405). There also is evidence that lactulose has prebiotic properties. However, lactulose induces osmotic effects in the intestine and acts as a laxative; consequently, it is not considered a useful food ingredient. There are various other candidate prebiotics for which evidence of prebiotic properties, and certainly of potential health benefits, needs to be demonstrated (isomalt, xylo-oligosaccharides, lactosucrose, soyabean oligosaccharides, isomalto-oligosaccharides, etc.).
Prebiotics and experimental models of inflammation
Inulin increased colonic lactobacilli and decreased cecal pH in rats subjected to DSS-induced intestinal inflammation(Reference Videla, Vilaseca and Antolin406). Inulin decreased the lesion score of the colonic mucosa, which correlated very well with the reduction in intraluminal release of inflammatory markers such as PGE2, TXB2 and LTB4 and reduction in mucosal myeloperoxidase activity, a marker of neutrophil accumulation within the tissue.
Schultz et al. (Reference Schultz, Munro and Tannock407) used two groups of HLA-B27 transgenic rats that spontaneously develop immune-mediated colitis and administered both a prebiotic (oligofructose) and probiotics (L. acidophilus and Bifidobacterium lactis). They observed a significant improvement of the histologic score, which was correlated to prebiotic effects on the ceco-colonic microflora. No probiotic bacterial strains were detected in the faeces. In a similar model, Hoentjen et al. (Reference Hoentjen, Welling and Harmsen408) administered a combination of rapidly and slowly fermented fructoligosaccharides and observed significant improvement of histologic score in the cecum and colon. The authors observed a concomitant improvement of inflammation markers such as a decrease in IL-1β and an increase in TGF-β in the cecal tissue, and a decrease in IFN-γ in the mesenteric lymph nodes. These effects again correlated with the impact of the dietary prebiotics on cecal microflora (increased lactobacilli and bifidobacteria), implying that the selective prebiotic interaction with the intestinal flora was the basis of the observations. A long-term (33 week) study of tumour formation and immune function in AOM-treated rats revealed that prebiotics (inulin), and a combination of prebiotics and probiotics, had different effects on the gut-associated lymphoid tissue than probiotics (Lactobacillus rhamnosus and B. lactis)(Reference Femia, Luceri and Dolara409).
Prebiotics and inflammatory conditions
Saavedra et al. (Reference Saavedra, Tschernia and Moore410) showed reduced absenteeism, visits to the physician and fever-associated diarrhoea in kindergarten-aged children given prebiotics. These results have been recently confirmed by Waligora-Dupriet(Reference Waligora-Dupriet, Campeotto and Nicolis411) in 6- to 24-month-old infants who were given a rapidly fermented prebiotic (oligofructose). Reduced flatulence and diarrhoea, as well as vomiting and fever – clinical observable consequences of inflammation – were correlated with the typical selective prebiotic modification of the intestinal flora (increased bifidobacteria and decreased C. perfringens and Staphylococcus spp.). In another study, a combination of galacto-oligosaccharides and long-chain inulin decreased the incidence of atopic dermatitis in babies at high risk(Reference Moro, Arslanoglu and Stahl412). This effect was accompanied by a significant modification of the composition of the intestinal microflora, characterised by higher Bifidobacterium spp. counts.
The anti-inflammatory effects of prebiotics have been investigated in patients suffering from inflammatory conditions. Welters et al. (Reference Welters, Heineman and Thunnissen413) administered prebiotic chicory inulin to familial adenomatous polyposis patients with ileal pouch-anal anastomosis. There was a significant decrease in the pouchitis disease activity index, confirmed by a decrease in endoscopic score, which correlated with a modified faecal short-chain fatty acid pattern (increased butyrate), which is a direct consequence of selective interaction of the administered prebiotics on intestinal flora. In an exploratory study, Casellas et al. (Reference Casellas, Borruel and Torrejon414) administered either oligofructose-enriched inulin or placebo (maltodextrin) to patients with active UC and on standard drug therapy (mesalazine). The prebiotic induced a faster reduction in faecal calprotectin levels than that seen in the control group. However, the rate of clinical remission was similar in both placebo and prebiotic groups, as all patients were on standard medical therapy with mesalazine. Furrie et al. (Reference Furrie, Macfarlane and Kennedy27) administered a synbiotic composed of oligofructose-enriched inulin as a prebiotic and Bifidobacterium longum as a probiotic to patients with UC. Sigmoidoscopy scores were reduced in the synbiotic group as compared with control. Mucosal mRNA levels for human β-defensins (hBD2, hBD3 and hBD4) that are upregulated in active UC were reduced in the synbiotic group as were inflammatory cytokines such as TNF-α and IL1-α. Biopsies in the synbiotic group had reduced inflammation and epithelial tissue was regenerated. These anti-inflammatory effects of the synbiotic combination were accompanied by a typical prebiotic-type shift in composition of the intestinal microflora, characterised by a 42-fold increase in numbers of Bifidobacterium spp. associated with the mucosa.
Probiotics
Description, sources, normal intakes and roles
The FAO of the UN (FAO) and the WHO have recognised the potential of probiotic foods to provide health benefits and the use of specific strains as safe supplements for human consumption. Probiotics are defined as ‘live micro-organisms, which, when administered in sufficient amounts, confer a health benefit to the host’(415). Lactic acid-producing bacteria, including lactobacilli and bifidobacteria from human origin, are the most commonly used probiotics. Foods containing probiotic bacteria range from dairy fermented milk products to probiotic powders, drinks or supplements in a lyophilised form. Probiotic effects can be direct or indirect through modulation of the endogenous intestinal microflora or immune system. Their fate in the gastrointestinal tract and their effects differ among strains. For most of the strains, the effective dose is between 108 and 1010 cfu/d. Over the past decade, progress has been made to provide scientifically and clinically valid evidence on the impact of defined probiotic strains on human health.
Mechanisms of anti-inflammatory action
Experimental models have revealed that probiotics differ greatly in their mechanism of action, including modification of the local microflora, epithelial barrier function, inflammation or the immune system. Several probiotic bacteria adhere to mucosal tissue in a strain-dependent manner, and by this transiently enhance intestinal persistence and limit pathogen access to the epithelium(Reference Bergonzelli, Granato and Pridmore416, Reference Buck, Altermann and Svingerud417). Modulation of the gastrointestinal microenvironment and inhibition of pathogen growth by probiotics was demonstrated via the release of antimicrobial factors, such as defensins, bacteriocins, hydrogen peroxide, nitric oxide, and SCFA and reduction in the luminal pH(Reference Penner, Fedorak and Madsen418). Several probiotics, such as L. acidophilus ATCC4356 or VSL#3, a mixture of four different lactobacilli, three bifidobacteria and Streptococcus thermophilus, have been shown to successfully preserve epithelial barrier function by induction of mucin secretion, maintenance of cytoskeletal integrity, tight junction protein phosphorylation or induction of heat-shock proteins(Reference Petrof, Kojima and Ropeleski419, Reference Cario, Gerken and Podolsky420). The anti-inflammatory effect of a number of probiotic bacteria, including lactobacilli and bifidobacteria, seems to be based on their properties to directly interact with intestinal epithelial cells, which have a key role in sensing danger signals within the luminal microenvironment. Some probiotic bacteria antagonise NF-κB activation by degradation of IκB-α, or regulating the nuclear export of NF-κB relA or NF-κB p65 subunits in a PPAR-γ-dependent manner(Reference Neish, Gewirtz and Zeng421, Reference Kelly, Campbell and King422). Also, expression of pro-inflammatory genes, such as TNF-α, IL-1 or neutrophil-recruiting and activating IL-8, which are transcriptionally regulated by NF-κB, was shown to be decreased by several probiotic strains, including VSL#3, Lactobacillus salivarius UCC118, Bifidobacterium infantis 35 624 and Lactobacillus johnsonii La1(Reference O'Hara, O'Regan and Fanning423). On the other hand, in a recent study, B. lactis Bb12 has been shown to target the toll-like receptor-2 signalling cascade, suggesting that probiotics themselves may trigger innate pro-inflammatory responses(Reference Ruiz, Hoffmann and Szcesny424). Similar pro-inflammatory effects by the activation of cytokines via toll-like receptor-2 are also reported for probiotic strains L. rhamnosus GG(Reference Miettinen, Lehtonen and Julkunen425) and L. casei Shirota(Reference Matsuguchi, Takagi and Matsuzaki426).
The induction of protective responses by probiotics is highlighted by the capacity of some strains (or their components) to interact with gut-residing DC, e.g. via DC-SIGN, to induce their maturation and predominant secretion of IL-10, which is thought to favour the induction of regulatory T-cells(Reference Smits, Engering and van der Kleij427). In addition to regulatory T-cells, TGF-β signalling in IECs plays a crucial role in maintaining mucosal immune homeostasis, as TGF-β regulates expression of MHC class II molecules and activity of metalloproteinases(Reference Hahm, Im and Lee428). Inhibition of TGF-β signalling due to overexpression of the Smad7 protein, a negative regulator of TGF-β, in IECs was shown to perpetuate the production of pro-inflammatory cytokines(Reference Monteleone, Kumberova and Croft429). VSL#3 improved Th1-mediated experimental murine colitis by restoring the cytokine balance through induction of IL-10 and TGF-β bearing regulatory T-cells(Reference Di Giocinto, Marinaro and Sanchez430). Also, increased TGF-β signalling induced by Bifidobacterium breve in preterm infants has been associated with reduced expression of Smad 7(Reference Fujii, Ohtsuka and Lee431).
Effects in inflammatory conditions
Inflammatory bowel diseases
A large body of evidence from both experimental animal work and clinical observations suggests that the enteric microflora may play an important role in driving the abnormal inflammatory response in the genetically susceptible host leading to chronic inflammation in IBD. This has led to the interest and increasing scientific evaluation of the use of probiotics as means to reconstitute microbial and immunological homeostasis. Although there are promising results for certain single probiotic strains in experimental models of colitis, the picture is more variable for the emerging human intervention studies using probiotic micro-organisms or combinations thereof as therapeutic interventions in IBD. Combinations of several probiotic strains have demonstrated efficacy in the control of pouchitis, an inflammation of the ileal-anal anastomosis, surgically produced after colorectomy for UC. In the present context, a randomised, placebo-controlled study with high dose of VSL#3 was shown to be effective in the maintenance of antibiotic-induced remission in chronic pouchitis(Reference Mimura, Rizzello and Helwig432). By contrast, the use of single probiotic strains, such as L. rhamnosus GG or a combination of L. acidophilus and B. lactis did not show clinical improvement in acute active pouchitis(Reference Kuisma, Mentula and Jarvinen433). In the treatment of acute CD, in a small number of children with mild to moderate disease activity, L. rhamnosus GG in combination with prednisone and immunomodulatory drugs showed clinical improvement(Reference Gupta, Andrew and Kirschner434). However, in adult patients with active CD L. rhamnosus GG was ineffective, although patients were on tapering steroid diet regimen and received antibiotics before starting the probiotic supplementation(Reference Schultz, Timmer and Herfarth435). In the maintenance of surgically-induced remission, a few studies with divergent outcomes are reported. In a randomised placebo-controlled prospective study, oral administration of L. rhamnosus GG for 1 year could neither prevent Crohn's recurrent lesions after surgery nor reduce their severity(Reference Prantera, Scribano and Falasco436). By contrast, administration of VSL#3 demonstrated efficacy in preventing post-operative recurrence when compared to mesalamine(Reference Gionchetti, Rizzello and Venturi31) up to 1-year post-surgery. Two recent studies have shown no benefit from L. johnsonii La1 supplementation for prophylaxis of post-operative recurrence of inflammatory lesions in adult CD patients(Reference Marteau, Lemann and Seksik437, Reference Van Gossum, Dewit and Louis438). The therapeutic efficacy of E. coli Nissle 1917 in maintaining remission in UC patients has been demonstrated in human trials (equally efficacious as the standard mesalazine), while the same probiotic treatment lacked convincing data in active disease(Reference Kruis, Fric and Pokrotnieks439, Reference Rembacken, Snelling and Hawkey440). A randomised, open-label, multicentre trial showed that VSL#3 in combination with low-dose balsalazide (a prodrug of mesalazine) was more effective than standard doses of basalazide or mesalazine monotherapy in the treatment of acute mild to moderate UC(Reference Tursi, Brandimarte and Giorgetti441). The efficacy of Lactobacillus GG in combination with mesazaline to sustain remission in patients with quiescent UC has been shown recently(Reference Zocco, dal Verme and Cremonini442). The role of bifidobacteria in medically induced remission of UC was evaluated in a small human trial showing less relapses in the bifidobacteria-supplemented patients during a 2-month follow-up. Inflammatory activity in the colon, assessed by NF-κB, IκB and pro-inflammatory cytokine analysis (IL-1β, TNF-α), was significantly decreased in the probiotic treatment group(Reference Cui, Chen and Wang443). Similarly, a fermented milk product with probiotic bifidobacteria had a positive effect on UC patients(Reference Ishikawa, Akedo and Umesaki444). Finally, a randomised controlled pilot trial with B. longum in combination with a inulin-type prebiotics showed resolution of inflammation in UC patients and reduced inflammatory activity in the mucosal tissue (less TNF-α, IL-1α, and defensins)(Reference Furrie, Macfarlane and Kennedy27).
Taken together, the human study results demonstrate that the therapeutic intervention with probiotics in IBD is encouraging, but not as straightforward as expected from experiences with experimental animal models of colitis. Single probiotic strains have been demonstrated to successfully prevent or treat experimental colitis. However, the overall picture of probiotic therapy in human IBD is by far more complex. To date only a few randomised, double-blind placebo-controlled trials are published, which (so far) impose the conclusion that high doses of probiotics and most likely a combination of different lactobacilli and bifidobacteria (or prebiotics) are more effective in decreasing clinical inflammatory score and maintaining patients in medically induced remission than a single probiotic strain. Future research might identify clinical phenotypes of IBD patients (based on insights in genetic predisposition, microbiota asset and immune response) that are likely to respond to probiotic therapy. As a result of the emerging understanding of microbial activities in different parts of the intestine (in health and disease) and increasing scientific evidence of probiotic mechanisms, an educated and targeted selection of specific probiotic strains and combinations thereof will be possible in the future to apply probiotic therapy in conjunction with medical treatments to maintain clinical remission.
Rheumatoid arthritis
The treatment of experimental adjuvant arthritis with a combination of methotrexate and lyophilised Enterococcus faecium enriched with organic selenium was shown to be beneficial in reducing inflammation (serum albumin, serum nitrite/nitrate concentrations, changes in hind paw swelling, arthrogram score) and bone erosion in rats(Reference Rovensky, Svik and Stancikova445). However, the probiotic alone was not effective. One small-scale human trial on L. rhamnosus GG in human RA did not show a clinical benefit(Reference Hatakka, Martio and Korpela446).
Atopic eczema
Studies with animal models reveal that several lactobacilli strains including plantarum and casei have the potential to inhibit pro-allergic immune responses(Reference Shida, Makino and Morishita447–Reference Shida, Takahashi and Iwadate450). Infants with atopy show a different profile of gut microflora than non-atopic controls(Reference Bjorksten, Naaber and Sepp64, Reference Björkstén, Sepp and Julge451) and a low number of bifidobacteria and lactobacilli predicted atopy development in Finnish infants(Reference Kalliomaki, Kirjavainen and Eerola396, Reference Ouwehand, Isolauri and He452). Studies in pregnant women and infants have demonstrated that probiotics (L. casei GG) lower the risk of development of atopic eczema in infants(Reference Ouwehand, Isolauri and He452–Reference Kalliomäki, Salminen and Arvilommi455), this effect being associated with lowered levels of inflammatory markers in faeces. Rosenfeldt et al. (Reference Rosenfeldt, Benfeldt and Nielsen457) showed that the combination of L. rhamnosus and L. reuteri reduced atopy severity and serum eosinophil cationic protein in 13-year-old atopic children.
CVD
Probiotic interventions in CVD are restricted to a few reports on modulation of risk factors associated with atherosclerosis, such as antioxidant defences or atherosclerotic lipid profiles. Hlivak et al. (Reference Hlivak, Odraska and Ferencik458) showed a reduction in LDL cholesterol after administration of Enterococcus faecium M-74 enriched with selenium. No significant alterations in HDL and TAG were observed. A second study reported a reduction in CVD risk factors (systolic blood pressure; leptin, fibrinogen, F(2)-isoprostanes, IL-6) in hyper-cholesterolaemic patients (heavy smokers) after consumption of L. plantarum 299v. Monocytes from the probiotic-treated group showed significantly reduced adhesion to native and stimulated human umbilical vein endothelial cells(Reference Naruszewicz, Johansson and Zapolska-Downar459).
Conclusions
Inflammation is a stereotypical physiological response to infections and tissue injury; it initiates pathogen killing as well as tissue repair processes and helps to restore homeostasis at infected or damaged sites. Acute inflammatory reactions are usually self-limiting and resolve rapidly. This involves the activation of negative feedback mechanisms such as the secretion of immunoregulatory cytokines (e.g. IL-10 and TGF-β), inhibition of pro-inflammatory signalling cascades, receptor shedding (e.g. TNF-R) and activation of regulatory cells. Thus, regulated inflammatory responses are essential to remain healthy and maintain homeostasis. However, inflammatory responses that fail to regulate themselves can become chronic and contribute to the perpetuation and progression of disease.
Characteristics typical of chronic inflammatory responses underlying the pathophysiology of several disorders include loss of barrier function, responsiveness to a normally benign stimulus, infiltration of inflammatory cells into compartments where they are not normally found in such high numbers, and overproduction of oxidants, cytokines, chemokines, eicosanoids and matrix metalloproteinases (Table 1). The levels of these mediators amplify the inflammatory response, are destructive and contribute to the clinical symptoms. Why resolution of inflammation is absent or abnormal in so many pathophysiological processes remains largely unknown.
Different factors including Western lifestyle (pollution, stress) and diet (low intake of fruits and vegetables, polyphenols and other antioxidants) have been postulated to reduce the efficiency of antioxidant defences, shifting the redox balance and thus increase the risk of inflammatory responses becoming chronic (Fig. 6). This may partially explain why many inflammatory disorders are more prevalent in Western countries and that in many cases prevalence increases with the level of westernisation(Reference Bach460).
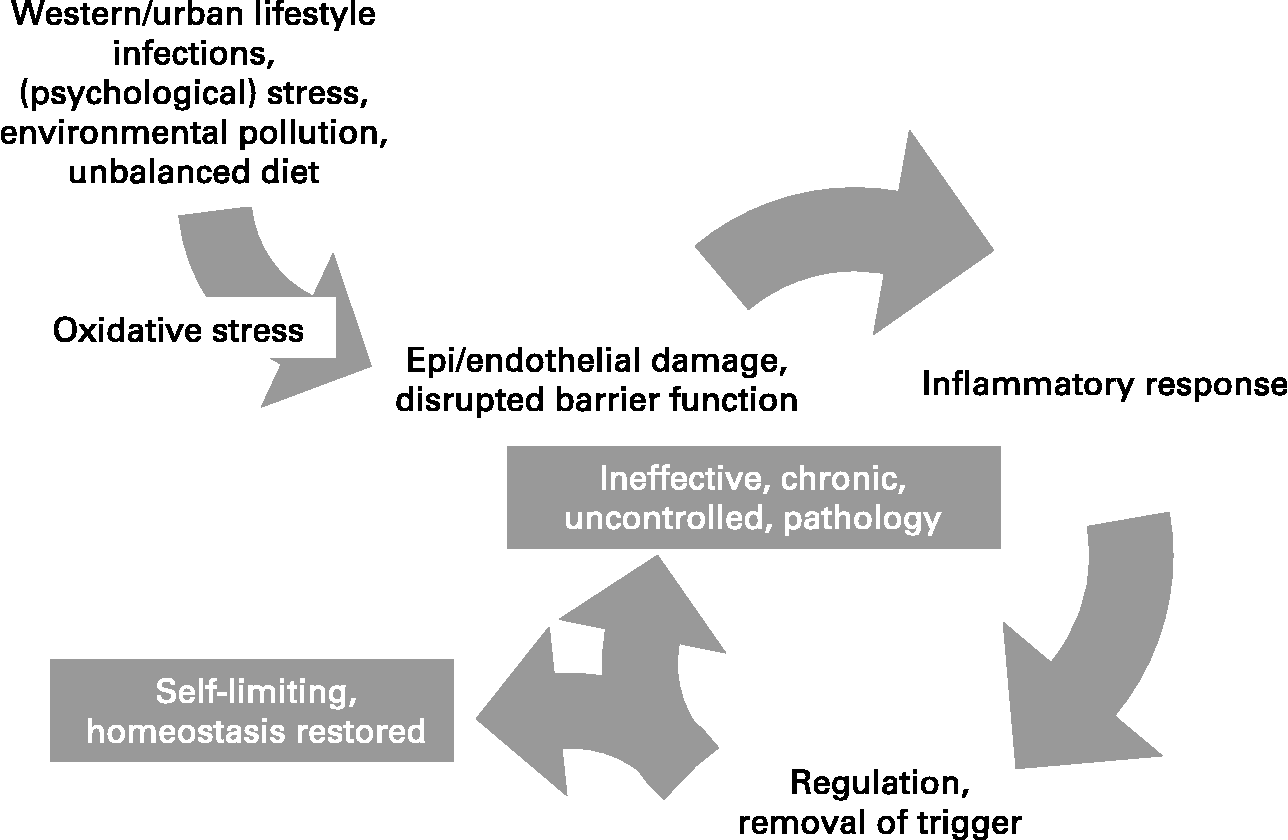
Fig. 6 Proposed link between Western lifestyle and chronic inflammation. Different factors associated with a Western lifestyle such as pollution, (psychological) stress and an unbalanced diet (low intake of fruits and vegetables, polyphenols and other antioxidants) reduce the efficiency of antioxidant defences, shifting the redox balance thus increasing the risk of inflammatory responses becoming chronic.
Various dietary components have been suggested to impact on inflammatory conditions, but the number of studies assessing therapeutic benefits of dietary interventions in established inflammatory disorders is still fairly limited. There is good evidence of efficacy of n-3 PUFA in RA with less strong evidence in CD and psoriasis and rather weak evidence in UC, asthma and eczema (Table 3). These fatty acids are also beneficial in established CVD, but the extent to which this is attributable to their anti-inflammatory effects is not clear. Dietary antioxidants represent a crucial line of defence against oxidative and inflammatory insult common to the development of many pathological disorders and a potentially protective role of dietary antioxidants in disease prevention is supported by much basic scientific evidence. The common mechanism of oxidative stress development in most of the here-described pathologies make the role of dietary antioxidants crucial for an optimal preventive action. Despite these considerations, trials in patients suggest little clinical benefit from antioxidant vitamins and polyphenolics in the disorders considered here, although there is some evidence for benefit from vitamin E in RA (Table 3). In order to optimise the levels of protection against oxidative and inflammatory stress, the concept of antioxidant efficiency should be re-evaluated investigating the synergistic interactions among components of the redox network of the body. Efforts should be focused on the identification of the composition of redox network and of the mechanism(s) underlying its homeostatic modulation. The link between inflammatory and antioxidant status needs to be more deeply investigated with well-tailored in vivo studies. Particular attention needs to be paid towards the interactions between the two systems in healthy subjects and during the different phases of the inflammatory response in the development of pathologies.
Table 3 Summary of the effects of VLC n-3 PUFA, antioxidant vitamins, flavonoids, prebiotics and probiotics on immune/inflammatory markers and conditions

Yes indicates an effect has been demonstrated; ?Yes indicates effects on a clinical condition have been shown but that these are not conclusive; ? indicates not known.
Th, helper T-cell; NK, natural killer; PMN, polymorphonuclear cell; COPD, chronic obstructive pulmonary disease.
The intestinal flora is in intimate contact with the most developed immunological organ of the human body imbedded in the intestinal membranes. There is a continuous intensive interaction established between the intestinal bacterial ecosystem and the host. The composition of this ecosystem can be improved either by selectively stimulating the resident bacterial populations that contribute to the protection against inflammatory processes (with prebiotics) or by adding external bacteria that are known to protect against inflammation (certain probiotics). Another important aspect is to locally stimulate the metabolic activity in a selective way (with prebiotics) so as to optimise the interaction with the immune system. There is evidence that consumption of prebiotics leads to clinical improvement in UC and CD, with limited or no evidence in the other conditions described here (Table 3). There is good evidence that probiotics bring about clinically relevant improvements in UC and CD and protect against development of atopic dermatitis in infancy (Table 3). There is little or no evidence of efficacy in the other conditions described here. It is possible that nutrition may have a bigger impact on prevention rather than treatment of chronic inflammatory conditions. Studies with different dietary components in various models and clinical settings have demonstrated that dietary compounds modulate pathways involved in controlling inflammation including intracellular signalling pathways, transcription factor activity and generation of inflammatory mediators (Table 3). Bioavailability and tissue accumulation of antioxidant vitamins and n-3 PUFA are not a problem, but low bioavailability of flavanoids and the lack of studies showing a specific accumulation of flavonoids in blood and target tissues raise concerns about their role as anti-inflammatory agents in vivo. More studies and a larger body of evidence are needed for these phytochemicals.
By reinforcing the regulatory pathways controlling inflammatory responses, nutrition may contribute to the robustness of homeostatic control and may help to reduce the risk that acute inflammatory responses, which initiate pathogen killing as well as tissue repair processes, derail into uncontrolled chronic inflammatory responses that contribute to perpetuation and progression of disease (Fig. 7). Nutrition may thus help to widen the boundaries within which the inflammatory responses can deal with the challenges posed by exposure to pathogens, allergens, toxins, tissue damage, etc., without spiralling into degenerative chronic inflammatory responses (Fig. 7). Among the dietary components considered here, long-chain n-3 PUFA, antioxidant vitamins, plant flavonoids, prebiotics and probiotics all have the potential to modulate predisposition to chronic inflammatory conditions and may have a role in their therapy.

Fig. 7 Concept of how nutrients might act in an anti-inflammatory manner. By reinforcing the regulatory pathways controlling inflammatory responses, nutrition may contribute to the robustness of homeostatic control and may help to reduce the risk that acute inflammatory responses persist as uncontrolled chronic inflammatory responses. Nutrition may thus help to widen the boundaries within which the inflammatory responses can deal with the challenges posed by exposure to pathogens, allergens, toxins, tissue damage, etc., without spiralling into degenerative chronic inflammatory responses.
There is a clear need for more good-quality studies assessing the impact of nutrition and specific nutrients on chronic inflammatory responses. Ideally, such studies should assess relevant clinical symptoms of chronic inflammation as well as selected markers of inflammation. Not only will this help to understand the underlying mechanism, but it will also help to predict which other inflammatory conditions may benefit from particular (combinations of) anti-inflammatory dietary compounds. The putative effect of nutritional compounds in strengthening the regulatory networks controlling inflammatory responses should be studied in more detail using conditions and models in which a stress is applied to the homeostatic control of inflammation.
Acknowledgements
This work was commissioned by the Nutrition and Immunity Task Force of the European branch of the International Life Sciences Institute (ILSI Europe). Industry members of this task force are: FrieslandCampina, Groupe Danone, Nestlé, Seven Seas, Südzucker/BENEO Group, Royal Cosun, Unilever, Valio, Yakult. The opinions expressed herein are those of the authors and do not necessarily represent the views of ILSI Europe.
Author contributions
Authors drafted sections of the review as follows: F. G. (Gut: Celiac disease), S. M. (Gut: Inflammatory bowel diseases), M. L. (Lungs: Airways and allergic inflammation), W. v. E. (Joints: Rheumatoid arthritis), P. S. F. (Skin: Atopic eczema and psoriasis), G. A. F. (The vascular wall: CVD), G. S. F. (Adipose tissue: Obesity), P. C. C. (Fatty acids), B. M. W.- R. (Antioxidant vitamins), M. S. (Flavonoids), J. v. L. (Prebiotics), J. Z. and S. B. (Probiotics). All authors contributed to discussions at the Workshop and had input into the final version of the review. P. C. C. chaired the workshop and led the writing of the review.
Conflict of interest statement
P. C. C. has research funding from Beneo-Orafti and B. Braun Melsungen, at the time of drafting the review was a consultant to Equazen and Royal Dutch Numico and has received speaking fees from Solvay Healthcare, B. Braun Melsungen, Fresenius Kabi and Seven Seas Limited. R. A. is an employee of Unilever. J.- M. A. and R. B.- S. are employees of Danone Vitapole. S. B. is an employee of Nestle. G. A. F. has received speaking fees from Pfizer, Fournier, Astra-Zeneca and Roche. G. F. is an employee of Curax. F. G. is a member of the Beneo Scientific Committee sponsored by Orafti and of the Cimizia Advisory Board sponsored by UCB Pharma. P. D. M. is an employee of Sensus. At the time of the Workshop, L. M. was an employee of Royal Numico, now known as Danone Research Centre for Specialised Nutrition. M. S. has research funding from Coca-Cola and BPW. J. v. L. is an employee of Südzucker/BENEO Group. W. V. D. is an employee of Seven Seas Limited. S. V. is an employee of ILSI Europe. B. M. W.- R. has a research collaboration with DSM Nutritional Products. J. Z. is an employee of Yakult. P. S. F., G. S. F., S. M. and W. v. E. have no conflicts of interest to declare.










