



Mammalian cells use myoinositol (Ins), a polyol that used also to be known as vitamin B8, as the core constituent of eight Ins phospholipids (the phosphoinositides, see Box 1), of many Ins polyphosphates and of the glycerophosphoinositol-based (GPI) anchors of cell surface proteins( Reference Michell 1 ) Footnote § . It is quite difficult to make mammals overtly Ins-deficient, except under nutritionally extreme conditions – and then a major symptom is fat accumulation in the liver and/or intestine. It is now commonly assumed that a normal diet plus some endogenous synthesis usually provides us (and other animals) with an adequate supply of Ins.
Box 1 Inositols and inositol phospholipids in eukaryotes: summary

However, recent evidence suggests that consuming extra Ins as a dietary supplement can be beneficial in a variety of human conditions, particularly those characterised by adiposity, hyperglycaemia and insulin resistance. These conditions include polycystic ovary syndrome (PCOS), gestational diabetes mellitus (GDM) and metabolic syndrome (MetS). Other possible benefits of Ins supplements include better sperm development and motility, improved ovarian follicle and embryo development, suppression of diabetes-induced and folate-resistant developmental birth defects, and amelioration of respiratory distress syndrome and retinopathy in premature infants (see references Reference Croze and Soulage2–Reference Greene, Leung and Copp5, for reviews). At least some of these suggested clinical benefits will probably become more firmly established once larger trials have been conducted.
The most prevalent hypothesis in discussions of these exciting observations has been that eating extra Ins might support the formation and actions of proposed, but controversial, ‘mediators’ of insulin action that contain inositols as constituents( Reference Croze and Soulage 2 , Reference Monastra, Unfer and Harrath 6 ). However, there is no direct evidence in support of this idea, and none of these discussions has attempted to explain how eating quite a lot of Ins – maybe enough to approximately double the usual dietary intake – might selectively amplify this minor and ill-understood facet of the Biology of Ins.
It seems more likely that the extra dietary Ins corrects a mild systemic Ins deficiency that restricts the production of an abundant and essential Ins-containing cell component, and the obvious candidate is phosphatidylinositol (PtdIns), a ubiquitous constituent of eukaryotic cell membranes. If this is correct then the central problem becomes to discover how allowing stressed tissues to make more PtdIns contributes to their health?
This review attempts to answer this question by:
(1) giving a snapshot of the diverse biological functions of Ins and its derivatives;
(2) summarising the endogenously synthesised and dietary sources of Ins;
(3) looking back to early work on Ins deficiency in mammals and considering how recent work on endoplasmic reticulum (ER) stress might help us to understand it better;
(4) summarising the evidence that Ins supplements ameliorate several conditions in which overweight, insulin resistance, hyperglycaemia and ER stress often coexist; and
(5) considering what mechanisms might underlie these benefits, and how experimentally to explore them.
These considerations lead to the conclusion that we should in future focus our attention on understanding when and why the ER sometimes needs to increase its production of PtdIns, the cell’s most abundant and least studied phosphoinositide, especially when a cell’s homeostatic adjustment to ER stress demands an increase in the rate of ER membrane synthesis – and how limitation of the Ins supply may prejudice this response.
Ins functions and supply
During the last four decades it has been recognised that PtdIns and its seven polyphosphorylated derivatives (the polyphosphoinositides (PPIn)) (see Box 1 and Fig. 1) have many essential functions, particularly in transmembrane signalling, in ‘labelling’ the identities of membrane compartments and in membrane trafficking( Reference Michell 1 , Reference Di Paolo and De Camilli 7 – Reference Hammond and Balla 10 ). Ins is also the precursor to a plethora of water-soluble Ins polyphosphates, only a few of which have understood functions( Reference Shears 11 – Reference Wilson, Livermore and Saiardi 14 ). And some PtdIns is channelled to the luminal face of the ER and incorporated into PtdIns glycan structures that will form the ‘GPI anchors’ that tether many proteins onto the external surfaces of eukaryotic cells( Reference Low 15 , Reference Zurzolo and Simons 16 ). Alongside these well-established functions there is 30 years of less than conclusive evidence, to be discussed below, that insulin receptor stimulation might activate a phospholipase that liberates inositol phosphate glycan molecule(s) (IPGs) similar to those in GPI anchors, and that these might serve as second messengers that initiate some tissue responses to insulin.
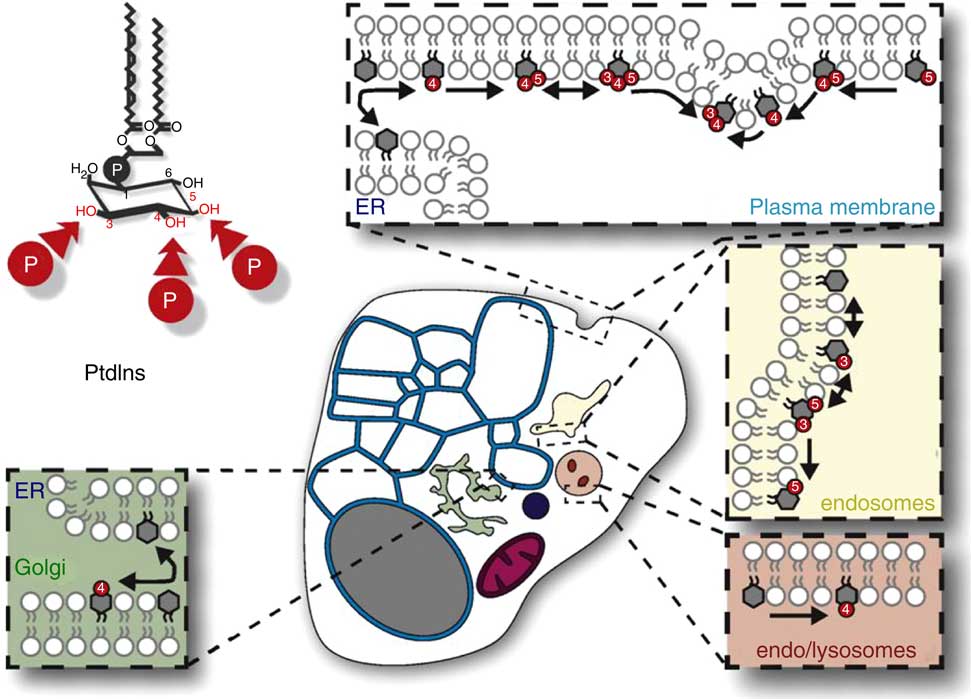
Fig. 1 The distribution, relative amounts and interconversions of Ins phospholipids in a generalized mammalian cell. This figure is reproduced, with permission, from reference (10), but the legend is mine. This generalised representation of a cell summarizes the distributions and relative quantities of the various organelles and of phosphatidylinositol (PtdIns) and the various polyphosphoinositides (PPIn) in these membrane subcompartments of eukaryote cells. The relative areas of membrane and volumes of the various organelle compartments, and thus their relative contents of membrane lipids (including PtdIns and PPIn) will vary substantially between cell-types. Important points to note include the following: (1) in many cells the endoplasmic reticulum (ER)/nuclear envelope continuum (the blue reticulum in this image) usually contains a larger proportion of a cell’s membranes than other organelle systems; (2) PtdIns is made in the ER (blue) and is the only phosphoinositide there (except maybe for transient traces of PtdIns4P); (3) PtdIns is a structural lipid that is distributed amongst all cell membranes (with less in the mitochondrial inner membrane), and is also the substrate for synthesis and turnover of all of the compartment-specific PPIn that are in the various non-ER and non-mitochondrial membrane systems (images of which are schematically expanded in the coloured boxes in this diagram).
Free Ins is the precursor of all of these molecules, and eukaryote cells get Ins from two sources. Some tissues, including brain and kidneys, and especially testes, make Ins de novo from the central metabolite d-glucose-6-phosphate by the sequential actions of myoinositol 3-phosphate synthase (MIPS, encoded in yeast by Ino1 and in humans by ISYNA1) and inositol monophosphatases (InsPases, encoded in yeast by Inm1 and Inm2, and in mammals by IMPA1 and IMPA2)( Reference Majumder, Chatterjee and Ghosh Dastidar 17 ). Secondly, specific cation/Ins co-transporters that use Na+ or H+ as co-solutes (SMIT1/SLC5A3, SMIT2 and HMIT/SLC2A13) harvest Ins from the gut, take it into the tissues from the blood and recover it from the renal glomerular filtrate( Reference Schneider 18 ). It is commonly assumed that cells use imported and locally made Ins in similar ways, but some observations suggest that this is not always the case( Reference Reynolds 19 , Reference González-Salgado, Steinmann and Major 20 ).
All eukaryotic cells need an Ins supply to make PtdIns and other Ins derivatives. It was discovered long ago that yeasts only survive if they can make and/or import Ins; that many mammalian cell-lines require an external Ins supply( Reference Eagle, Oyama and Levy 21 – Reference Henry, Gaspar and Jesch 23 ); and that mammalian embryos which cannot make Ins develop abnormally, and the offspring die perinatally unless they are fed Ins from birth( Reference Ohnishi, Murata and Watanabe 24 ).
Dietary Ins comes mainly from the PtdIns in plant and animal foodstuffs and from the inositol polyphosphates (mainly phytic acid; InsP 6) that are abundant in many seeds. Normal human Ins intake has been estimated to be in the range 0·25–2 g/d( Reference Clements and Darnell 25 , Reference Wells 26 ), but we lack precise estimates of how much of that dietary Ins we absorb. Taking InsP 6 as an example, different raw, cooked and processed foods contain very different amounts, and it is not even agreed how much of the Ins in ingested InsP 6 is absorbed and in what form( Reference Schlemmer, Frølich and Prieto 27 , Reference Dinicola, Minini and Unfer 28 ). Recent studies suggest that the gut cannot absorb intact InsP 6 ( Reference Wilson, Bulley and Pisani 29 ), so it is likely that dietary InsP 6 must be fully dephosphorylated before Ins transporters can absorb it from the gut.
Given this background, it is somewhat surprising that a growing body of information – which is discussed below and summarised in Table 1 – suggests that some people get substantial health benefits if they supplement their normal diets with an extra 1–4 g of Ins/d (see references Reference Croze and Soulage2–Reference Greene, Leung and Copp5, Reference Beemster, Groenen and Steegers-Theunissen30–Reference Muscogiuri, Palomba and Laganà32, for reviews). The most notable effects include an amelioration of insulin resistance and of the related metabolic problems that characterise PCOS, GDM, MetS and Type 2 diabetes mellitus (DM2). The text that follows: (a) briefly summarises a number of the studies that suggest that dietary supplementation with Ins is beneficial to humans and to relevant rodent models; (b) discusses the suggested interpretations of these studies; (c) offers a novel interpretation of these striking findings; and (d) suggests some experimental approaches that might help us to understand these findings and to explain the health benefits of this inexpensive and safe nutritional supplement.
First, though, three other topics need to be briefly discussed as background: a possible reinterpretation of studies – both older and more recent – of Ins deficiency in mammals and other metazoans; the roles of various Ins derivatives in insulin signalling; and the long history of relationships between diabetes and Ins metabolism.
Ins deficiency provokes the development of steatoses (and ER stress?)
One of the most notable early observations was that dietary Ins deficiency – usually in rats or gerbils – causes an Ins-reversible fat accumulation (steatosis) in the liver and/or the intestinal mucosa. This is most striking when withdrawal of Ins from the diet is combined either with a diet rich in saturated fat or with the nutrient stress of lactation (when the mother is secreting lots of Ins into her milk for the suckling offspring)( Reference Holub 22 , Reference Wells 26 , Reference Chu and Geyer 33 ). Dietary Ins supplementation sometimes also ameliorates fatty livers caused by other metabolic stresses, such as choline deficiency( Reference Wells 26 ). One unifying defect in these steatoses is a failure by the Ins-deficient organs efficiently to package and secrete the TAG-rich lipoproteins that they make (VLDL in hepatocytes and chylomicra in enterocytes). It has been assumed that a cellular deficit of PtdIns (and maybe of PtdIns-derived PPIn) somehow causes these symptoms – and during Ins deficiency in gerbils the PtdIns content of microsomal membranes from the intestinal mucosa was Ins-reversibly decreased by more than one-half( Reference Chu, Geyer and Walker 34 ).
The underlying mechanisms of these steatoses have never been fully understood, and by the late 1980s there was a widespread belief that a combination of endogenous Ins synthesis and a balanced diet provides an adequate supply of Ins to most mammals, including humans( Reference Croze and Soulage 2 , Reference Holub 22 , Reference Clements and Darnell 25 , Reference Wells 26 ). As a result, most researchers turned their attention to problems that seemed more exciting.
More recently, it has been recognised that transient or sustained hepatic steatosis – which may or may not indicate incipient or overt pathology – occurs in many situations of metabolic imbalance, and this has taken centre stage under a new name: non-alcoholic fatty liver disease (NAFLD)( Reference Henkel and Green 35 , Reference Lonardo, Ballestri and Marchesini 36 ). A variety of metabolic stresses can provoke and/or are closely associated with NAFLD (or TAFLD, its Toxicant-Associated equivalent( Reference Wahlang, Beier and Clair 37 )). These conditions include central obesity, saturated fat feeding and viral hepatitis, as well as toxic insults such as carbon tetrachloride poisoning or tunicamycin inhibition of protein glycosylation. NAFLD is common in patients with several of the conditions for which dietary Ins supplements are now promoted, including PCOS, GDM, MetS and DM2( Reference Lonardo, Ballestri and Marchesini 36 , Reference Bruce and Byrne 38 – Reference Ajmera, Gunderson and Van Wagner 40 ).
NAFLD is one of the commonest hepatic accompaniments of the widespread unfolded protein response (UPR) that is evoked in all eukaryotes by diverse stressors that cause accumulation of incompletely folded proteins within the ER – a situation that is known generically as ‘ER stress’. Cellular responses to ER stress, which in the liver often include fat accumulation in hepatocytes, are orchestrated by a complex set of coordinated controls, the master regulators of which are three transmembrane ER proteins: IRE1 (Inositol-Requiring Enzyme-1), which is present in all eukaryotes (mammals have two: IRE1α and IRE1β); PERK (PKR-like ER Kinase), which is restricted to metazoans; and ATF6 (Activating Transcription Factor-6), which is also restricted to metazoans (mammals have two: ATF6α and ATF6β) (see references Reference Ron and Walter41–Reference Ariyasu, Yoshida and Hasegawa47 and the later discussion). The ubiquitous ER stress sensor IRE1 is a dual-activity protein kinase and endoribonuclease that is central to responses provoked by diverse stressors, but its name is a confusing hangover from the fact that it was first identified as a key player in the responses of yeast to changes in external Ins supply( Reference Henry, Gaspar and Jesch 23 , Reference Nikawa and Yamashita 48 ).
Three more recent studies have shown that interfering with the PtdIns supply in cells in other ways can provoke steatoses that have some similarities to those seen in classical studies of Ins deficiency – and which again resemble the NAFLD that is associated with ER stress and the UPR.
First, Alb et al. ( Reference Alb, Cortese and Phillips 49 , Reference Alb, Phillips and Wilfley 50 ) observed that reducing or abolishing rats’ ability to make PITPα, a cytosolic phospholipid exchange protein that may have a role in distributing PtdIns around cells, provokes hepatic and intestinal steatoses similar to those provoked by Ins deficiency( Reference Nile, Bankaitis and Grabon 51 ).
Secondly, ablation in zebrafish embryos of the gene encoding PtdIns synthase (PIS, also known as CDP-diacylglycerol-inositol phosphotransferase (CDIPT)) is lethal in 5–6 days. But these PIS-null embryos develop a fatty liver during the final developmental days before they die. It seems that they initially import maternally provided PtdIns from yolk, and this allows them to develop fairly normally for up to ~4 days, but when this runs out they briefly develop an acute PtdIns deficiency and a fatty liver – in which a suite of genes indicative of ER stress is turned on – and then they die. And treatment of wild-type embryos with tunicamycin, an inhibitor that induces ER stress by preventing the glycosylation and correct folding of nascent proteins within the ER, produces remarkably similar effects( Reference Thakur, Stuckenholz and Rivera 52 ). These PIS-null embryos also develop an intestinal mucosal pathology that partially mimics inflammatory bowel disease, and which appears to be initiated by disruption of the ER architecture in enterocytes( Reference Thakur, Davison and Stuckenholz 53 ).
Third, Drosophila larvae that lack or have reduced levels of PIS or of CDP-diacylglycerol synthase (which makes the immediate lipid precursor of PtdIns) develop only partially and are non-viable. Many of their PtdIns-deprived tissues stay abnormally small during their abortive development and display abnormal intracellular accumulations of fat droplets( Reference Liu, Wang and Shui 54 ).
Considered as a whole, these results suggest that: (a) the liver and intestinal steatoses that were recognised many decades ago as hallmarks of Ins deficiency were early examples of NAFLD and its intestinal equivalent; and (b) these steatoses were probably unrecognised consequences of the fact that a cellular deficit of Ins, and thus of PtdIns, stresses the ER and activates the protective/homeostatic ER stress pathways. These steatoses are particularly striking accompaniments of ER stress in hepatocytes and enterocytes, both of which are cells whose ER’s major tasks include the synthesis and assembly of secretory lipoproteins, but the Drosophila observations also suggest that the stress of PtdIns depletion may provoke fatty infiltration of other tissues( Reference Liu, Wang and Shui 54 ). And the Ins-reversible loss in Ins-deficient gerbils( Reference Chu, Geyer and Walker 34 ) of much of the enterocyte’s alkaline phosphatase – a GPI-anchored protein that is normally made by the ER and trafficked to the exterior of the brush border membrane that faces the gut lumen – was probably another early indication of ER malfunction provoked by PtdIns deficiency.
It therefore seems likely that ER stress is widespread in the tissues of overtly Ins-deficient animals, especially in those tissues in which cells are subjected to fluctuating demands for protein secretion and/or for ER expansion (such as the insulin-secreting β-cells of pancreatic islets of Langerhans( Reference Thomas, Dalton and Daly 55 )). The final sections of this review will suggest a novel explanation of the benefits of dietary Ins supplements that is based on the idea: that the ER must manufacture new membrane phospholipids, including PtdIns, whenever a cell needs to expand its ER membrane area as an element of its response to ER stress; that a mild global Ins deficiency often interferes with this process in metabolic diseases such as PCOS and GDM; and that Ins supplements can relieve this deficiency.
Ins derivatives in insulin signalling, and the inositol derivatives used as dietary supplements
Insulin resistance is one of the physiological features of many of the conditions for which dietary Ins supplements are being promoted( Reference Croze and Soulage 2 ); and Ins-containing molecules have at least one, and maybe two or three, role(s) in insulin signalling.
First, the predominant mode of insulin receptor signalling into cells is via receptor-catalysed tyrosine phosphorylation of insulin receptor substrate-1 (IRS-1), which then activates Type I PPIn 3-kinase (PI3K-I) (and other targets). Activated PI3K-I converts a small amount of plasma membrane PtdIns(4,5)P 2 into PtdIns(3,4,5)P 3, and this PtdIns(3,4,5)P 3 recruits and regulates multiple effector proteins that control diverse cell responses (reviewed in reference Reference Cantley56).
Secondly, soluble Ins-containing second messengers (or ‘mediators’) might mediate some of the actions of insulin. A decade before insulin-stimulated PI3K-I signalling was discovered, Joseph Larner’s laboratory offered evidence that insulin-stimulated skeletal muscle makes a soluble ‘mediator’ of insulin action( Reference Larner, Galasko and Cheng 57 ). Subsequent work has suggested that: (a) two such mediators (named IPG-A and IPG-B) might each control a different subset of tissue-specific responses to insulin; (b) each IPG contains an inositol (myoinositol in IPG-A and D-chiro-inositol (DCI) in IPG-B); and (c) these IPGs are structurally related to, and might be derived from, the GPI anchors of cell surface proteins( Reference Larner, Brautigan and Thorner 58 – Reference Suzuki, Suzuki and Hinokio 60 ). However, different laboratories have reported very different biological effects even when working with similar molecules( Reference Hecht, Tsai and Liu 59 , Reference Suzuki, Suzuki and Hinokio 60 ), and the status of these ideas remains uncertain after almost 40 years.
Despite these uncertainties, the idea that eating substantial dietary supplements of Ins may augment insulin signalling through IPGs has dominated many recent discussions( Reference Croze and Soulage 2 , Reference Noventa, Vitagliano and Quaranta 4 , Reference Monastra, Unfer and Harrath 6 ). This emphasis has been especially prevalent in studies that have focussed on supplements consisting of Ins/DCI combinations, of DCI alone or of plant-derived DCI derivatives such as pinitol (3-O-methyl-DCI). DCI differs from myoinositol only by inversion of the configuration of one hydroxyl group on the inositol ring, and DCI is normally present in mammalian blood and urine, though at much lower concentrations than Ins( Reference Hong, Jang and Kang 61 ). Much of this DCI comes from the diet, and it still not clear whether mammals make DCI from Ins. Ins and DCI have been reported to be interconvertible through the action of an partially characterised epimerase activity that is found in mammalian tissues( Reference Larner, Brautigan and Thorner 58 , Reference Unfer, Carlomagno and Papaleo 62 ), but a study of intact rodents found no evidence for their interconversion in vivo ( Reference Lin, Ma and Gopalan 63 ).
Some workers promote the virtues of dietary Ins supplements that contain DCI( Reference Monastra, Unfer and Harrath 6 ), others are non-committal( Reference Galletta, Grasso and Vaiarelli 64 , Reference Facchinetti, Bizzarri and Benvenga 65 ) and some cite their disadvantages( Reference Garg and Tal 66 ). Given the uncertainties about the Biology of DCI – its interconvertibility (or not) with Ins, its role (or not) in insulin signalling and its value (or not) as a supplement – the remainder of this discussion will be confined to exploration of possible ways in which dietary supplements of Ins, a molecule that has many undisputed and well-understood roles in cell function, might help to alleviate the effects of metabolic and hormonal imbalances. DCI and/or 3-O-methyl-DCI undoubtedly have interesting biological effects, including improved diabetic control in Type II diabetics( Reference Kim, Kim and Kang 67 ), neuroprotection in diabetic mice( Reference Farias, Macêdo and Oquendo 68 ), suppression of inflammation in a mouse asthma model( Reference Lee, Lee and Jeong 69 ), prolongation of fruit-fly lifespan( Reference Hada, Yoo and Seong 70 ) and very rapid activation of PKB/Akt in cultured endothelial cells( Reference D’Oria, Laviola and Giorgino 71 ), but we still have little idea how these are brought about.
Ins and diabetes
It has been known since the 19th century that excess fluid intake, including that occurring in the various types of diabetes, causes polyuria and accelerates Ins loss into urine( Reference Needham 72 ), and Daughaday et al.( Reference Daughaday and Larner 73 , Reference Daughaday, Larner and Houghton 74 ) established long ago that elevated blood glucose competes with inositol for renal reabsorbtion. They concluded
‘that the inosituria of diabetes mellitis can be attributed to an increase in the renal clearance of inositol produced by glycosuria’.
This focussed attention on the idea that, secondary to this urinary loss, diabetic people and animals might suffer from an Ins deficiency that would contribute to the pathogenesis of at least some of the disease’s long-term consequences, such as neuropathy, nephropathy and retinopathy. Moreover, diabetic mothers, both human and rodent, are at increased risk of producing offspring with neural crest-related malformations, and by 2000 a substantial body of work suggested that a modest Ins deficiency might cause some of these developmental defects( Reference Eriksson, Borg and Cederberg 75 ).
The second route by which the body loses Ins is through its oxidation to glucuronate by proximal tubular myoinositol oxidase in the kidney cortex, with the product metabolised via the glucuronate/xylulose pathway. This route of Ins loss is also over-active in diabetes, hypertension and obesity – and in response to raised plasma insulin concentration, as is typically seen in conditions involving insulin resistance( Reference Chang, Chao and Walker 76 – Reference Tominaga, Dutta and Joladarashi 78 ).
The consequence of these adverse renal events is that any person or animal that is diabetic and/or insulin-resistant, and so is likely to be hyperglycaemic and/or to have elevated circulating insulin, will lose Ins by one or both of these routes and will be at increased risk of systemic Ins deficiency. The idea that Ins deficiency may be an important feature of diabetes has therefore become an enduring theme, though without an agreed explanation of how it brings about its effects( Reference Croze and Soulage 2 ).
Conditions for which dietary Ins supplements are therapeutic
Published reports on the effects of Ins supplements have fairly consistently reported them to be helpful, based on evidence of variable quality, for a substantial range of human medical conditions and in animal models of these conditions. Table 1 briefly summarises the most convincing of these observations, most of which have been discussed in detail by the reviews cited in the table legend. The condition-specific references within the table include more recent primary sources and also meta-analyses and reviews of earlier studies. A few of the latter are Cochrane Collaboration reviews of the possible clinical benefits of Ins supplements: for example, in preventing or alleviating GDM and in supporting the normal development of premature babies.
Several of the conditions that benefit from Ins supplements – including PCOS, GDM, MetS and T2D – involve related metabolic and endocrine derangements and tend to share physiological characteristics such as central obesity, insulin resistance, NAFLD and tissue inflammation. These, in turn, are increasingly being linked at a cellular level with the phenomenon of ER stress in the most affected tissues( Reference Thomas, Dalton and Daly 55 , Reference Liong and Lappas 79 – Reference Bañuls, Rovira-Llopis and Martinez de Marañon 82 ). Other conditions listed in Table 1 are less obviously related, but ER stress contributes to at least some aspects of the pathologies involved in retinopathy( Reference Zhang, Ma and Bhatta 83 ), hypothyroidism( Reference Zhou, Ding and Li 84 ), diabetic embryopathy( Reference Wang, Weng and Quon 85 ) and defective spermatogenesis( Reference Guzel, Arlier and Guzeloglu-Kayisli 86 ).
The beneficial studies listed in Table employed supplements of no more than 4 g Ins per day, without any recorded adverse effects. Moreover, some other trials, mainly in anxiety and depressive disorders, have administered much larger doses of Ins, typically 12–18 g/d, and have observed neither significant side-effects nor appreciable clinical benefit( Reference Carlomagno and Unfer 87 , Reference Mukai, Kishi and Matsuda 88 ).
Many of the benefits of inositol supplements that are listed in Table 1 have been observed both in human patients and in more rigorously controlled studies of animal models. Not surprisingly, few of the human studies have been large enough and/or of sufficiently rigorous design to yield unambiguous outcomes that could justify firm clinical recommendations, even when combined in meta-analyses. As a result, the relevant meta-analyses have often concluded with some variant of ‘Encouraging, but more studies are needed in larger and more diverse populations’.
We also should not ignore the possibility that studies that remain unpublished, which would be more likely to be negative or inconclusive, might cause some publication bias. For example, the open invitation to submit papers for a 2016/2017 themed collection entitled ‘Inositol(s) from Bench to Bedside in Endocrinology and Gynecology’ in the International Journal of Endocrinology stated that ‘inositols play a pivotal role, as drugs (my emphasis), in treating several pathologies such as PCOS, MetS and GDM’ (https://www.hindawi.com/journals/ije/si/828614/cfp/). The invitation solicited papers on any aspect of inositols as dietary therapies, but did not suggest that this collection might also have served as an appropriate home for inconclusive or negative findings.
The animal studies have often yielded much clearer evidence of benefits of Ins feeding observed under well-controlled laboratory conditions – for example, in decreasing the prevalence of neural tube defects (NTDs) in the progeny of ‘curly tail’ mice that are genetically prone to develop folate-resistant NTDs( Reference Greene, Leung and Copp 5 , Reference Cockroft, Brook and Copp 89 , Reference Greene and Copp 90 ).
Might all benefits of Ins supplements rely on a single mechanism?
What cellular mechanism(s) underlie the beneficial effects of substantial Ins supplements on the conditions listed in Table 1? Possible explanations are of two types. On the one hand, they might involve multiple effects, with the benefit to each condition having its own individual explanation, as has sometimes been hypothesised (see in next paragraph). Alternatively, it seems more likely that one essential, but still unidentified, set of Ins-dependent cell functions operates less than optimally in all of the Ins-responsive conditions, and that Ins supplements supply extra Ins to support this function.
Several specific effects of the former type have been postulated: these include augmentation of insulin signalling through IPGs (see above), effects on cytoskeletal regulation( Reference Bizzarri, Cucina and Dinicola 91 ), phosphorylation events downstream of phospholipase C signalling( Reference Cogram, Hynes and Dunlevy 92 ) and inositols acting as antioxidants that protect against the reactive oxygen species generated by cell metabolism( Reference Artini, Casarosa and Carletti 93 ). Several of these would selectively implicate different subsets of the many cell regulation processes that depend on Ins derivatives of low abundance (such as the various PPIn, and maybe IPGs) – but none of these reports has ever, to my knowledge, made any attempt to explain how a modest global Ins deficiency might bring about such selective actions. Indeed, it is hard to imagine how the effects of doubling a person’s dietary Ins intake could be targetted at improving metabolic support for any of the various pathways that rely on, or are regulated by, rare Ins-containing cell constituent(s) – and that different faults in such events could contribute selectively to different maladies.
It seems far more likely that there is a single, common, explanation – i.e. that generous Ins supplements ensure that cells always have enough Ins to sustain some central cell processes that rely on an abundant Ins-containing cell constituent. If that is the case, we must answer two questions: (a) which abundant Ins-containing molecule has its supply maintained by the Ins supplements; and (b) what central cell processes are compromised by limiting the supply of this molecule, especially during metabolic and endocrine imbalances?
The remainder of this review will outline an hypothesis that attempts to answer these questions. It will: (a) suggest that metabolically stressed tissues develop a mild Ins deficiency, and that consuming extra Ins supports the increased PtdIns biosynthesis that is needed whenever stressed ER needs to increase its functional capacity and enlarge its membrane area; and (b) discuss how experimentally to test this novel interpretation of how extra dietary Ins improves ER function in stressed cells.
Ins supplements ensure a sustained supply of PtdIns – but for what purpose?
PtdIns is the main Ins-containing membrane phospholipid of all eukaryote cells (see below). It is much more abundant than any of a cell’s other Ins-containing constituents and is the precursor of most of them. Any substantial decrease in the availability of Ins is therefore likely to diminish a cell’s PtdIns supply (see Box 1 and the text below).
However, we know much less about specific functions of PtdIns – assuming there are some? – than we should. This is at least partly because the discovery of the PtdIns(4,5)P 2-dependent phosphoinositidase C signalling pathway in the early 1980s shifted the main focus of the phosphoinositide field away from PtdIns (and the ER) onto PtdIns(4,5)P 2 (and the PM). And workers in this field have since been kept fully occupied by an unending programme of identifying the various other PPIn and decoding their many functions (at various non-ER sites) (as outlined in Fig. 1). As a result, it seems likely that we have been unfairly neglecting the possibility, and maybe likelihood, that PtdIns, a major membrane constituent, has specific roles in membrane function and dynamics that are still to be identified.
One useful outcome of trying to understand the beneficial effects of dietary Ins supplements may be that it makes us pose two important questions( Reference Michell 1 , Reference Michell 94 ). First, ‘Why do cells make so much more PtdIns than they would need if PtdIns only served as a precursor to other Ins-containing cell constituents?’ And maybe more fundamentally, Why have phospholipids with an Ins1P headgroup remained an essential part of the cellular apparatus of Archaea and their Eukaryotic descendants throughout most of the time that there has been life on Earth?
For eukaryotes like us, the most frequently offered answer would still probably be ‘PtdIns is the essential precursor from which all of the PPIn are made, so we need plenty of it’. However, this answer won’t wash for archaea, because the membranes of most of these diverse organisms contain archaetidylinositol (ArcIns: sn-2,-3-dialkyl-glycero-1-phospho-1D-myoinositol) or a close relative, but probably have no phosphorylated ArcInsP n derivatives analogous to the PPIn of eukaryotes (except for ArcIns4P as an intermediate in ArcIns biosynthesis( Reference Morii, Kiyonari and Ishino 95 )). This surely makes it more likely that phospholipids with an unadorned Ins1P headgroup (ArcIns in archaea, and PtdIns in eukaryotes and a few bacteria) first evolved in archaea to fulfil some fundamental and ubiquitous biological function(s) – which we do not yet understand – and that eukaryote cells have both retained this (or these) function(s) and elaborated many more?( Reference Michell 1 , Reference Michell 94 )
How much PtdIns, where in the cell is it and how is it made?
How much PtdIns do cells contain and how is it distributed amongst their membrane systems? There are three key variables for each type of tissue or cell – the total PtdIns concentration, the PtdIns concentration in each organelle and the relative membrane masses of the various organelle systems.
Mammalian cells are typically quoted as having ~10 % of their membrane phospholipid as PtdIns( Reference Vance 96 ). PIS has a relatively low affinity for Ins, so the main factor that controls the rate of PtdIns synthesis is the cytosolic free Ins concentration rather than any regulated change in intrinsic PIS activity. PtdIns is ~12–27 % of the total phospholipid in Saccharomyces cerevisiae, with the higher figure from yeast grown with abundant exogenous inositol; and when yeast’s capacity for PtdIns synthesis is varied genetically it maintains normal growth rates with as little as 4 % PtdIns, but can only grow slowly with 2 % PtdIns( Reference Jani and Lopes 97 ).
Older estimates on mammalian tissues showed total lipid-bound Ins concentrations of ~2–3 mm in brain (ox and guinea-pig) and liver (rat), with almost all of it in PtdIns( Reference Dawson and Eichberg 98 , Reference Michell, Hawthorne and Coleman 99 ). A recent phosphoinositide analysis of mouse tissues found similar PtdIns concentrations and also determined the relative concentrations of PtdIns, of PtdInsP and PtdInsP 2 (their isomers were not resolved) and of PtdInsP 3. PtdIns was again predominant: it constituted more than 98 % of the total in liver and about 92 % in brain( Reference Anderson, Kielkowska and Durrant 100 ).
A lipidomic analysis of membranes from cultured RAW264.7 macrophages probably offers the most authoritative information on PtdIns concentrations in organelles: it found slightly more PtdIns per mg membrane protein in the PM than the ER, with considerably less in mitochondria( Reference Andreyev, Fahy and Guan 101 ). An analysis of cultured mouse embryo fibroblasts also suggested modest PtdIns enrichment in the PM relative to the ER( Reference Shulga, Myers and Ivanova 102 ).
Many years ago I measured the distribution of PtdIns amongst well characterised primary cell fractions from rat liver (unpublished results). Ins was acid-liberated from dried lipid extracts, trimethylsilylated and assayed by GLC. About half of the PtdIns was in a microsomal fraction that was ~3-fold enriched in the ER marker enzyme glucose 6-phosphatase and contained about one-fifth of the sedimentable 60 % of the homogenate protein. There was little PtdIns in a fraction consisting largely of mitochondria (~5-fold enriched in succinate dehydrogenase).
Table 1 The benefits of ins supplements in human patients and in animal models of human disease

GDM, gestational diabetes mellitus; IVF, in vitro fertilization; N/A, no assessment available; DCI, D-chiro-inositol.
In most of the human studies subjects consumed 1–4 g of extra Ins per day, which is a readily tolerated amount (see the text), but none of these studies include information that permit judgments about the relative merits of different quantities. Ins was taken in addition to routine folate in most studies of pregnancy (human and rodent). Some of the Ins supplements also contained a small amount of DCI (typical Ins:DCI ratio 40:1, when present), but the available information allows no conclusions about whether DCI significantly changed the effects of the supplements (see the text). For reasons of space, this table mainly cites recent focussed reviews, recent primary articles and/or meta-analyses – apologies to the authors of earlier primary sources who are not cited as a result. Many of the human and animal studies have been summarised in broader reviews( Reference Croze and Soulage 2 , Reference Noventa, Vitagliano and Quaranta 4 , Reference Greene, Leung and Copp 5 , Reference Beemster, Groenen and Steegers-Theunissen 30 – Reference Muscogiuri, Palomba and Laganà 32 , Reference Paul, Laganà and Maniglio 156 , Reference Pundir, Psaroudakis and Savnur 157 ). Bold reference numbers with asterisks (*) refer to Cochrane Collaboration systematic reviews.
This information suggests that PtdIns is distributed fairly evenly amongst a cell’s membranes, except for a considerably lower concentration in mitochondrial inner membranes( Reference Vance 96 ). In many cells, therefore, the bulk of the PtdIns will be in the pervasive tubuloreticular membrane network that makes up the ER, the cell’s most extensive membrane system( Reference Westrate, Lee and Prinz 103 , Reference Nixon-Abell, Obara and Weigel 104 ) (as illustrated in Fig. 1). This is also where phosphatidate (PtdOH) – either synthesised de novo or made by diacylglycerol kinase from phosphoinositidase C-liberated sn-1,2-diacylglycerol – is converted to PtdIns by a two-step pathway. First, cytidine diphosphate diacylglycerol (CDP-DG) synthase(s) (CDS1 and/or CDS2 in mammals) converts PtdOH to CDP-DG; and then a single PIS (PIS/CDIPT) transfers a phosphatidyl grouping from CDP-DG to the D-1-hydroxyl group of Ins. CDS1, CDS2 and PIS are all integral ER membrane proteins that have their active sites oriented towards the cytoplasm( Reference Waugh, Minogue and Clayton 105 – Reference Bochud and Conzelmann 107 ). There is growing evidence that there are important spatial and temporal controls over the non-homogeneous distribution of PtdIns and its biosynthesis within the ER( Reference Kim, Guzman-Hernandez and Balla 108 – Reference Bahmanyar 112 ), but most of this discussion will ignore these important details and consider the ER as a single extensive membrane entity that makes PtdIns and other membrane lipids for use by all of the cell’s membranes.
The majority of the PtdIns and PPIn molecules in many or most mammalian cells have, for ill-understood reasons, a remarkably homogenous fatty acyl composition, with a C18 : 0 (stearoyl) group on the sn-1 position of the glycerol backbone and a C20 : 4 (arachidonyl) group on the sn-2 position( Reference D’Souza and Epand 113 ). Much of the PtdIns that PIS makes is first synthesised with other fatty acid complements and is then remodelled by a phospholipase/acyltransferase cycle in which an arachidonyl-selective 2-acyltransferase (lysoPtdIns acyltransferase-1 (LPIAT1); also known as MBOAT7) is a major participant. LPIAT1 is essential both for mice to develop normally and for their PtdIns to achieve its distinctive fatty acid pairing( Reference Lee, Inoue and Sasaki 114 ). The tissues of the short-lived mice that lack LPIAT1 contain much less PtdIns than normal animals and their PtdIns is arachidonyl-depleted. In humans, inactivating mutations in LPIAT1/MBOAT7 give rise to a severe neurodevelopmental syndrome characterised by intellectual disability, autism and epilepsy( Reference Johansen, Rosti and Musaev 115 ), and humans carrying LPIAT1 allele rs641738, which has reduced expression, have a predisposition to liver steatosis, inflammation and necrosis( Reference Mancina, Dongiovanni and Petta 116 – Reference Eslam, Valenti and Romeo 119 ).
It therefore seems that if mammals are to survive, be healthy and have a normal PtdIns complement, they must be able correctly to make and to remodel a cytoplasm-facing ER pool of PtdIns that has several functions: (a) it exports PtdIns to the cytoplasmic leaflets of the cell’s other membranes, where some cycles rapidly in and out of small metabolic pools of the cell’s seven organelle-specific PPIn (see Box 1 and Fig. 1); (b) some of the PtdIns flips to face the ER lumen, where its fatty acid pattern is remodelled and it is incorporated into GPI anchors that will moor proteins to the exterior of the PM( Reference Zurzolo and Simons 16 , Reference Kinoshita, Maeda and Fujita 120 ); and (c) it contributes about one-tenth of the phospholipid that makes up the lipid bilayer in the ER membrane, so we must assume that more PtdIns must be made whenever the ER needs to be enlarged.
The UPR and ER membrane expansion: two faces of cell responses to ER stress
The remainder of this discussion will focus on the importance of maintaining an adequate PtdIns supply in order to support ER membrane homeostasis. The events by which cells respond to ER stress have been studied in exquisite detail in the yeast S. cerevisiae, much less in cultured mammalian cells and – despite the mounting evidence that perturbation of ER homeostasis is an important feature of many metabolic and endocrine disorders – only to a limited extent in intact mammals( Reference Pagliassotti 45 – Reference Ariyasu, Yoshida and Hasegawa 47 ). Moreover, it is increasingly being recognised that the suite of cellular adaptive events that generally sails under the ‘ER stress’ flag is simply an extreme version of a set of processes by which cells constantly modulate their everyday functions as they adapt to fluctuating nutrient supplies, diurnal bodily rhythms, and etc.( Reference Walter and Ron 44 , Reference Chedid and Nair 121 – Reference Soeda, Cordero and Li 124 ).
The original yeast experiments revealed the key features of a set of archetypal ER stress responses in which activation of a single transmembrane ER protein (IRE1) coordinates a suite of events in which:
(1) the complex UPR apparatus identifies and disposes of aberrant newly synthesised proteins, regulates the ER’s ongoing protein load, and orchestrates a suite of genes whose products enhance the ER’s luminal capacity for post-translational processing, folding and quality control of proteins;
(2) the ER expands its membrane area, a process that demands accelerated synthesis of phospholipids to enlarge the membrane bilayer, and this expansion both increases the internal volume within the ER for protein processing and allows the membranes of the ER to accommodate more intrinsic proteins without the lipid bilayer becoming over-crowded; or
(3) the ER does both of the above in a coordinated manner (reviewed in references Reference Henry, Gaspar and Jesch23,Reference Ron and Walter41,Reference Mori42).
In mammalian cells, the various elements of the ER homeostatic responses are regulated in more complex, and sometimes tissue-specific, ways by the interplay of five transmembrane regulators (IRE1α, IRE1β, PERK, ATF6α and ATF6β) and the proteins with which they functionally interact( Reference Ron and Walter 41 , Reference Mori 42 , Reference Walter and Ron 44 , Reference Fagone and Jackowski 125 ).
This discussion will focus on the membrane expansion arm of this dichotomy, since this is the process that particularly requires a coordinated increase in the synthesis of all of the phospholipids, including PtdIns, that make up the lipid bilayer of ER (and other) membranes.
The UPR, which is the ER’s response to protein misfolding within its lumen, is mainly initiated through interactions between any aberrant proteins that accumulate within the ER lumen and the luminal domains of these transmembrane regulators, especially IRE1α ( Reference Ron and Walter 41 , Reference Mori 42 ). It was therefore a surprise when it was discovered that truncated transmembrane versions of IRE1α and ATF6α that lack these luminal sensing domains can retain their ability to initiate dramatic increases in ER membrane biosynthesis. Occasionally this membrane expansion has been assessed directly (e.g. by fluorescence microscopy of cells expressing tagged variants of ER-resident proteins or by examination of thin-section electron micrographs( Reference Snapp, Hegde and Francolini 126 – Reference Christodoulou, Santarella-Mellwig and Santama 128 )) but more often it is detected less directly as an increase in the expression and/or activity of phospholipid-synthesising enzymes – usually CTP:phosphocholine cytidylyltransferase, the rate-limiting enzyme in phosphatidylcholine (PtdCho) synthesis by the Kennedy pathway( Reference Henry, Gaspar and Jesch 23 , Reference Fagone and Jackowski 125 , Reference Sriburi, Bommiasamy and Buldak 129 – Reference Lagace and Ridgway 131 ).
Effective inducers of such membrane expansion include exposure to exogenous saturated fatty acids (e.g. palmitate, C16 : 0), inhibition of the desaturation of endogenous stearate (C18 : 0) and the over-expression of intrinsic ER proteins – or of shortened constructs thereof that include little more than the relevant protein’s membrane-spanning domain. It therefore seems that the bilayer-spanning parts of the ER stress sensors (or of their membrane-spanning intrinsic ER partner proteins( Reference Covino, Ballweg and Stordeur 132 )) somehow sense an excessive degree of physical order within the membrane bilayer – caused, for example, by protein overcrowding or by a relatively saturated lipid environment – and compensate by initiating synthesis of the extra phospholipid that is needed to expand the area of ER lipid bilayer in which the proteins reside( Reference Takewaka, Zimmer and Hirata 133 – Reference Halbleib, Pesek and Covino 139 ). Remarkably, a recent study of variant IRE1α constructs showed that major changes can be made to the amino-acid sequence of this protein’s transmembrane domain without substantially changing its ability to initiate this response( Reference Kono, Amin-Wetzel and Ron 140 ).
Most efforts to explain control of the extra phospholipid synthesis that is needed to support ER membrane expansion have focussed, both in yeast and in mammalian cells, on defining how these ER stress sensors stimulate extra synthesis of the PtdCho that makes up about half of the phospholipid of the ER’s lipid bilayer( Reference Vance 96 , Reference Fagone and Jackowski 125 , Reference Lagace and Ridgway 131 , Reference Holthuis and Menon 141 ). Much less in known about the relative importance of phosphatidylethanolamine (PtdEtn), the second most abundant phospholipid of the ER, and of phosphatidylserine (PtdSer) and PtdIns, the other invariant and essential ER membrane phospholipids, and of how their biosyntheses are regulated( Reference Birner, Bürgermeister and Schneiter 142 ). But one defined consequence of genetically limiting liver cells’ ability to make either PtdEtn or PtdSer is to provoke hepatic ER stress, especially when the resulting PtdCho:PtdEtn ratio in the liver, which is normally around 1·6, substantially deviates to higher or lower values( Reference Van der Veen, Kennelly and Wan 143 ). Ongoing synthesis of these glycerophospholipids by the ER must, of course, also maintain an adequate supply of these membrane lipids to all of the cells’ other membranes, and the complex set of distribution processes by which this is achieved is finally beginning to be understood( Reference Holthuis and Menon 141 , Reference Van der Veen, Kennelly and Wan 143 – Reference Wong, Čopič and Levine 145 ).
How might we explore the role of PtdIns in ER function in mammalian cells and in animals?
It was yeast’s requirement for Ins as the essential PtdIns precursor that gave the name IRE1 to the prototypic ER stress sensor of yeast. The effects of limiting the supply of exogenous Ins to growing yeast are many, including: induction of the expression of Ino1, and thus of endogenous Ins production; a substantial decrease in the steady-state PtdIns concentration; a compensatory slowing of the synthesis of other membrane phospholipids, notably PtdCho, that are needed for membrane maintenance and expansion; and delaying the synthesis and trafficking of the GPI anchors of proteins destined for the PM. Overall, changing the availability of exogenous Ins influences the expression of at least 700 yeast genes( Reference Henry, Gaspar and Jesch 23 ).
The arguments presented above suggest that the most likely interpretation of the beneficial effects of dietary Ins supplements on various metabolic and endocrine imbalances in mammals is that they correct an Ins deficiency that prevents stressed ER from making enough PtdIns. So how can that idea be experimentally tested – ideally both in isolated and/or cultured cells and in intact animals? During recent decades we have been inundated by a flood of discoveries of ‘new’ and essential cell functions that rely on molecules made from PtdIns, especially the various PPIn, and this means that deciphering any specific functions of PtdIns will not be easy.
Analysis of the many functions of the PPIn has been facilitated by the development of an ingenious armoury of specific cell-biological tools that allow us to follow their individual distributions and movements in intact cells and to manipulate their concentrations enzymatically at localised intracellular sites( Reference Di Paolo and De Camilli 7 – Reference Hammond and Balla 10 ). However, there are not yet equivalent tools of high specificity with which to explore the functions of PtdIns – or, indeed, of any of the other abundant glycerophospholipids (PtdCho, PtdEtn and PtdSer) that, in various mixtures, dominate the core lipid bilayers of eukaryote cell membranes.
For example, we largely lack data, equivalent to those from yeast, on the effects of changing PtdIns-synthetic capacity or the availability of exogenous Ins on ER homeostasis in mammalian cells. Early studies established how much exogenous Ins various cultured cells need to support maximal growth, usually 1–3 µm, but nothing more( Reference Eagle, Oyama and Levy 21 ). And when, more recently, cultured cells have been radiolabelled for metabolic studies, this has generally been done at the lowest possible – and least costly – concentration that would kept them healthy (or at least ‘growing at an unrestrained rate under the conditions chosen for the experiment’). For example, ~5 µm Ins was the lowest concentration at which the HL60 promyeloid cells that we studied grew exponentially and appeared to differentiate normally towards neutrophils or monocytes( Reference French, Bunce and Stephens 146 ).
Unfortunately, such measurements tell us nothing about whether this would have remained an adequate Ins supply if these cultured cells had been subjected to the sorts of sustained demand for phospholipid biosynthesis that are seen during extensive ER membrane proliferation events: for example, (a) during the differentiation of immature B-lymphocytes, which have little ER, into ER-packed and Ig-secreting plasma cells( Reference Fagone, Sriburi and Ward-Chapman 147 , Reference Kirk, Cliff and Thomas 148 ); or (b) in response to provocations such as the various artificial membrane stresses mentioned above. Studies of the Ins requirements of – and the effects of Ins deprivation on – cells undergoing such adaptations should be very informative. Might Ins restriction limit their ER expansion and provoke overt ER stress, and maybe cause them to develop neutral lipid inclusions? Might the cells even bud off inflammation-provoking membrane vesicles in a manner similar to stressed hepatocytes( Reference Kakazu, Mauer and Yin 149 ) – and/or undergo stress-induced apoptosis?
Another ER stressor is high-fat feeding, which has long been known to potentiate the development of an Ins-responsive fatty liver in Ins-deprived animals( Reference Holub 22 , Reference Wells 26 ). When diabetes-prone mice were fed an extreme version of a high-fat diet for 1–4 months they developed a model of Type 2 diabetes that included tissue Ins depletion, inosituria and insulin resistance, some of which were partly reversed by feeding of large Ins supplements( Reference Croze, Géloën and Soulage 150 ).
Remembering that the nutrient drain of lactation was another potentiator of classical Ins deficiency-triggered liver steatosis (see earlier)( Reference Wells 26 ), one might also ask how limiting Ins supply would influence the mammary epithelium’s pre-partum differentiation in preparation for the start of lactation, a process in which the IRE1 effector XBP1 is now known to play an essential role( Reference Davis, Giesy and Long 151 , Reference Hasegawa, Calvo and Avivar-Valderas 152 ). It is known that women affected by GDM, a condition for which dietary Ins seems to be beneficial (see Table 1), tend to breastfeed less successfully than non-GDM mothers( Reference Oza-Frank and Gunderson 153 ). This is potentially detrimental both to the infant’s nutrition and to the subsequent health of the mother( Reference Gunderson, Hurston and Ning 154 ). Might continuing to take Ins supplements during lactation improve this situation by enhancing mammary function and Ins-enriching the mother’s milk – and thus help to sustain breast-feeding and improve the offspring’s nutrition? And might it also improve the mother’s chance of avoiding continued diabetes after her pregnancy?
In a broader context, one of the first things that might be done in a revival of ‘old-fashioned’ studies of rats, mice or gerbils would be to test the simple prediction that making these animals Ins-deficient would provoke many of their tissues to display diagnostic biochemical features of ER stress, and that these might be detectable well before the development of more overt features such as a fatty liver.
Finally, what do we know about the spatial organisation of the metabolism of PtdIns within the complex and ever-changing topology of the ER; what might its heterogenous distribution mean; and how might it be perturbed experimentally? First, it has recently been becoming clear that PIS, and thus PtdIns synthesis, tends to be concentrated in some peripheral elements of the ER, particularly where tubulovesicular elements make close contacts with other organelles( Reference Kim, Guzman-Hernandez and Balla 108 – Reference English and Voeltz 111 ) – although it will usually still be assumed that the new PtdIns equilibrates rapidly throughout the cytoplasmic surface of interconnecting ER membranes.
A hint that this may not always be true comes from an intriguing study of the role of the nuclear envelope protein CNEP-1 (C-terminal domain Nuclear Envelope Phosphatase-1), an activator of the phosphatidate (PtdOH) phosphatase known as lipin, in regulating nuclear membrane dynamics in Caenorhabditis elegans embryos. As illustrated in Fig. 2, the activity of this widely conserved regulator of substrate partitioning during glycerophospholipid synthesis somehow ensures that ectopic sheets of ER do not surround the nucleus and interfere with the normal dynamics of nuclear envelope disassembly during mitosis. The activated PtdOH phosphatase directs biosynthetic flux away from CDP-DG and PtdIns, and so dictates that the nuclear envelope lipid bilayer contains less PtdIns than the ’bulk’ ER. Confirmation that this relative nuclear depletion of PtdIns contributes to the sub-compartmentation of ER membrane distribution comes from the fact that global inhibition of PtdIns production can counteract the accumulation of these interfering perinuclear ER structures even when CNEP-1 is inhibited( Reference Bahmanyar 112 , Reference Bahmanyar, Biggs and Schuh 155 ). How this happens remains a mystery.
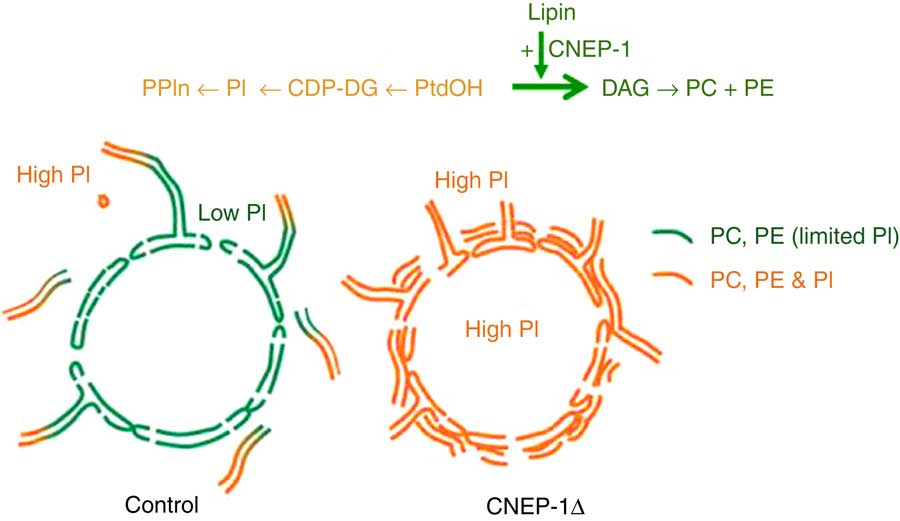
Fig. 2 Redirection of phospholipid synthesis by activation of phosphatidate (PtdOH) phosphatase causes the nuclear envelope to be relatively phosphatidylinositol (PtdIns)-depleted compared with the extra-nuclear endoplasmic reticulum (ER). The nuclear envelope-resident protein C-terminal domain Nuclear Envelope Phosphatase-1 (CNEP-1) activates lipin, a PtdOH phosphohydrolase, on membrane structures involved in nuclear envelope breakdown during cell division, so favouring the conversion of PtdOH to sn-1,2-diacylglycerol (DAG) over formation of cytidine diphosphate diacylglycerol (CDP-DG) (see upper pathway). As a result, the balance of phospholipid synthesis in these membranes is skewed away from PtdIns (PI) and towards PtdCho (PC) and PtdEtn (PE). The nuclear envelope therefore has a lower PtdIns content than the remainder of the ER continuum, and this somehow ensures that obstructive ectopic ER cisternae do not accumulate around the nucleus (lower schematic diagram). This figure is adapted, with permission, from Figure 1 of reference (112).
Conclusion
The observations discussed here suggest, rather surprisingly, that eating some daily inositol might make a simple dietary contribution to alleviating several common disorders. Drug companies have spent many years and many fortunes trying to find novel and costly treatments for these conditions, and the resulting drugs sometimes have unpleasant side-effects – whereas eating some extra Ins, a normal and inexpensive dietary constituent, seems benign. However, it remains worrying that well-validated explanations for its beneficial effects still elude us. Maybe the ideas put forward here might help to get us on the right track?
Acknowledgements
Several colleagues have offered valuable comments on these ideas as they have been developing. My particular thanks to Tamas Balla, Clifford Bailey, Gerry Hammond, Yusuf Hannun, Phill Hawkins, Esther Oppenheim and Len Stephens.
This research received no specific grant from any funding agency or from any commercial or not-for-profit sectors.
The author declares that there are no conflicts of interest.



