


Inflammatory bowel disease (IBD) is a widespread and debilitating illness characterised by the destruction of the gut mucosa by the mucosal immune system(Reference Strober, Fuss and Blumberg1). Activation of PPARγ has shown efficacy in the prevention or amelioration of experimental IBD(Reference Katayama, Wada and Nakajima2–Reference Lytle, Tod and Vo4). Results of a recent clinical study in ulcerative colitis patients demonstrate that rosiglitazone (Avandia™; GlaxoSmithKline, London, UK), an agonist of PPARγ and a Food and Drug Administration-approved drug for treating type 2 diabetes, is also efficacious in the treatment of mild-to-moderately active ulcerative colitis(Reference Lewis, Lichtenstein and Deren5). In spite of its efficacy, rosiglitazone is unlikely to be adopted for treating IBD due to its significant side effects(Reference Marcy, Britton and Blevins6, Reference Nesto, Bell and Bonow7) and a Food and Drug Administration-issued ‘black box’ warning and subsequent restriction of its use. Thus, there is a need to discover novel dual or pan-agonists of PPAR that exert therapeutic and prophylactic actions against IBD with limited or no adverse side effects.
A safer alternative to rosiglitazone in particular, or the thiazolidinedione class of anti-diabetic drugs in general (i.e. rosiglitazone, ciglitazone, troglitazone and pioglitazone), is conjugated linoleic acid (CLA), a naturally occurring fatty acid that ameliorates IBD through a PPARγ-dependent mechanism(Reference Bassaganya-Riera, Reynolds and Martino-Catt3, Reference Hontecillas, Wannemeulher and Zimmerman8, Reference Bassaganya-Riera and Hontecillas9). The efficacy of CLA against experimental IBD heightened our interest in discovering naturally occurring, orally active agonists of PPAR. In this regard, conjugated linolenic acids such as punicic acid (PUA), catalpic acid and eleostearic acid have demonstrated some promising effects as dual or pan-agonists of PPAR(Reference Bassaganya-Riera, Reynolds and Martino-Catt3, Reference Hontecillas, Diguardo and Duran10, Reference Hontecillas, O'Shea and Einerhand11). PUA is naturally found at high concentrations in the seed of Punica granatum (Punicaceae, pomegranate)(Reference Sassano, Sanderson and Franx12) amounting to 64–83 % of the pomegranate seed fatty acids(Reference Ahlers, Dennison and O'Neill13, Reference Kaufman and Wiesman14). Structurally, PUA is a conjugated octadecatrienoic acid containing c9, t11, c13 double bonds, resembling the c9, t11-CLA isomer – the predominant isomer in milk and beef(Reference Bassaganya-Riera, Hontecillas and Beitz15, Reference O'Shea, Bassaganya-Riera and Mohede16).
As with thiazolidinedione, PUA ameliorates glucose tolerance and obesity-related inflammation in animal models of obesity and type 2 diabetes by acting as dual PPARα and γ agonists(Reference Hontecillas, O'Shea and Einerhand11, Reference McFarlin, Strohacker and Kueht17, Reference Arao, Wang and Inoue18) with no adverse side effects detected in toxicological studies(Reference Meerts, Verspeek-Rip and Buskens19). At the gastrointestinal tract, PUA inhibits TNF-α-induced neutrophil hyperactivation, protects from experimental colitis(Reference Boussetta, Raad and Letteron20) and ameliorates inflammation-induced colorectal cancer(Reference Kohno, Suzuki and Yasui21). While some progress has been made in characterising some of the health effects of PUA, its underlying mechanisms of action are incompletely understood. The present study aims to elucidate the underlying mechanisms by which PUA ameliorates experimental IBD. Particularly, we investigate the role of PPAR as putative molecular targets for the prevention of IBD by PUA.
Experimental methods
Animal procedures
C57BL/6J wild-type mice and IL-10− / − mice were purchased from the Jackson Laboratories (Bar Harbor, ME, USA). Tissue-specific PPARγ null mice were generated as described previously(Reference Bassaganya-Riera, Reynolds and Martino-Catt3, Reference Akiyama, Sakai and Lambert22, Reference Wagner, McAllister and Ward23). The tail and colonic genotypes of mice were determined by PCR analysis as described previously(Reference Bassaganya-Riera, Reynolds and Martino-Catt3, Reference Cui, Miyoshi and Claudio24). PPARγ fl/fl; mouse mammary tumour virus (MMTV)-Cre+; IL-10− / − double knockout (DK) mice were generated by breeding IL-10− / − mice and tissue-specific PPARγ null mice expressing the MMTV-Cre recombinase in epithelial and haematopoietic cells. PPARδ null mice were backcrossed nine times to a C57BL/6J background and genotyped as described previously(Reference Barak, Liao and He25). We also utilised Villin-Cre mice lacking PPARγ in intestinal epithelial cells (IEC)(Reference Adachi, Kurotani and Morimura26, Reference Mohapatra, Guri and Climent27), CD4-Cre mice lacking PPARγ in T-cells(Reference Wohlfert, Nichols and Nevius28) and Lysozyme M-Cre mice lacking PPARγ in macrophages and neutrophils(Reference Shah, Morimura and Gonzalez29, Reference Hontecillas, Horne and Guri30). All mouse strains were bred under a C57BL/6J background. While we attempted to generate PPARδ; IL-10− / − DK mice, we were not successful due to increased embryonic mortality of mice lacking both PPARδ and IL-10. Mice were maintained at the experimental facilities at Virginia Polytechnic Institute and State University. All experimental protocols were approved by the Institutional Animal Care and Use Committee.
Dietary treatments and development of experimental colitis
Mice were fed purified diets for 6 weeks that represented a modification of the AIN-93G diet (Table 1) commonly used for the growth, pregnancy and lactational phases of mice(Reference Reeves, Nielsen and Fahey31). The optimal doses of PUA included in these diets were the result of time course and dose titration studies designed to elucidate the optimal anti-inflammatory efficacy of PUA performed previously (data not shown). Diets were prepared on a weekly basis, and feed was replaced on a daily basis to minimise fatty acid oxidation. Stock fatty acid solutions, e.g. pomegranate seed oil, were nitrogen-purged every time that the bottles were opened. The fatty acid profile of pomegranate seed oil was determined by NMR as described previously(Reference Sassano, Sanderson and Franx12) and shown to contain over 71 % PUA. For studies using IL-10− / − mice, breeder pairs were maintained in specific pathogen-free conditions, and pups were transferred into a conventional environment at weaning (21 d of age) to facilitate a greater microbial exposure and the development of experimental IBD. The treatment groups were (1) wild-type mice (negative control for colitis; n 12), (2) IL-10− / − mice with severe colitis at the start of the experiment (n 20) to investigate therapeutic efficacy, (3) IL-10− / − mice that have not developed colitis at the start of the experiment (n 40) to investigate prophylactic efficacy and (4) PPARγ fl/fl; MMTV-Cre+; IL-10− / − DK mice (n 20) to investigate the role of PPARγ in mediating the anti-inflammatory effect of PUA in the IL-10 model of spontaneous pan-enteritis. In the dextran sodium sulphate (DSS) studies, we used the following mouse genotypes: wild-type; whole-body PPARδ null; IEC-specific PPARγ null (Villin-Cre); macrophage-specific PPARγ null (Lysozyme M-Cre). Experimental diets provided a dose of PUA equivalent to 45–80 mg PUA/d per mouse. Subsequent studies used the DSS model of experimental IBD by inducing colitis by challenging mice with 2·5 % DSS, 36 000–44 000 molecular weight (ICN Biomedicals, Aurora, OH, USA) in the drinking-water for 7 d as described previously(Reference Bassaganya-Riera, Reynolds and Martino-Catt3).
Table 1 Composition of the diets*
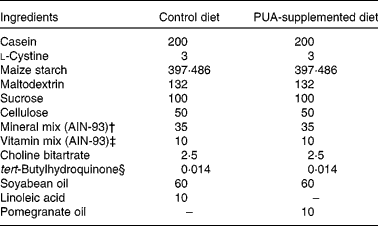
PUA, punicic acid.
* Provides approximately 7 % fat and 0·02 total cholesterol, and obtains 14·5 % of energy from fat. All dietary ingredients were purchased from Harlan Teklad (Madison, WI, USA), with the exception of pomegranate seed oil that was provided by Lipid Nutrition BV (Wormerveer, The Netherlands).
† Supplied per kg of diet: calcium carbonate, 357 g; potassium phosphate monobasic, 196 g; potassium citrate, 70·78 g; NaCl, 74 g; potassium sulphate, 46·6 g; magnesium oxide, 24·3 g; ferric citrate, 6·06 g; zinc carbonate, 1·65 g; manganous carbonate, 0·63 g; cupric carbonate, 0·31 g; potassium iodate, 0·01 g; sodium selenate, 0·01025 g; ammonium paramolybdate, 0·00795 g; sodium meta-silicate, 1·45 g; chromium potassium sulphate, 0·275 g; lithium chloride, 0·0174 g; boric acid, 0·0815 g; sodium fluoride, 0·0635 g; nickel carbonate, hydroxide, tetrahydrate, 0·0318 g; ammonium vanadate, 0·0066 g; sucrose, 220·716 g.
‡ Supplied per kg of diet: nicotinic acid, 3 g; calcium pantotenate, 1·6 g; pyridoxine HCl, 0·7 g; thiamin HCl, 0·6 g; riboflavin, 0·6 g; folic acid, 0·2 g; d-biotin, 0·02 g; vitamin B12 (0·1 % in mannitol), 2·5 g; dl-α-tocopheryl acetate (333·5 mg/g), 15 g; vitamin A palmitate (150 000 μg retinol/g), 0·8 g; vitamin D3 (cholecalciferol, 12 500 μg/g), 0·2 g; vitamin K (phylloquinone), 0·075 g; sucrose, 974·705 g.
§ Antioxidant.
Assessment of colitis
Mice were weighed on a daily basis and examined for clinical signs of disease associated with colitis (i.e. perianal soiling, rectal bleeding, rectal prolapses, diarrhoea and piloerection) by blinded observers. Disease activity indices were calculated using a modification of a previously published compounded clinical score(Reference Bassaganya-Riera, Reynolds and Martino-Catt3, Reference Saubermann, Beck and De Jong32). Briefly, disease activity index consisted of a scoring for diarrhoea and lethargy (0–3), whereas rectal bleeding consisted of a visual observation of blood in faeces and the perianal area (0–4). Results from preliminary studies demonstrated a high correlation between the results of faecal blood by Hemoccult and visual observations performed by experienced veterinarians. Mice in the DSS study were euthanised by CO2 narcosis followed by secondary thoracotomy on day 7 of the DSS challenge.
Histopathology
Segments of the colon (3 cm of the anatomic middle of the colon) were fixed in 10 % buffered neutral formalin, later embedded in paraffin, and then sectioned (6 μm) and stained with haematoxylin and eosin for histological examination. Tissue slides were examined as described previously(Reference Bassaganya-Riera, Reynolds and Martino-Catt3, Reference Bassaganya-Riera, Ferrer and Casagran33, Reference Hontecillas and Bassaganya-Riera34). Briefly, colons were graded with a compounded histological score including the extent of (1) crypt damage, (2) regeneration, (3) metaplasia/hyperplasia, (4) lamina proprial vascular changes, (5) submucosal changes and (6) presence of inflammatory infiltrates. The sections were graded with a range from 0 to 4 for each of the previous categories, and data were analysed as a normalised compounded score. We show the colonic results because the colonic lesions are common in the IL-10− / − and DSS colitis models. The ileal lesions can only be found in the IL-10− / − model but not in the DSS model, which is colon-specific.
Quantitative real-time RT-PCR from the colon
Total RNA was isolated from colonic samples using the RNeasy isolation kit (Qiagen, Valencia, CA, USA) to examine the expression of the three PPAR isoforms and PPAR-responsive genes. The PCR primer pairs for the genes of interest were designed based on previously published sequences (GenBank) using the Oligo 6 primer design software (Molecular Biology Insights, Cascade, CO, USA), and real-time RT-PCR was performed as described previously(Reference Bassaganya-Riera, Reynolds and Martino-Catt3, Reference Bassaganya-Riera and Hontecillas9). Briefly, total RNA was isolated from the whole colon of mice using the RNA isolation MiniKit (Qiagen) according to the manufacturer's instructions. All RNA samples were checked for quality and quantity on the Agilent 2100 BioAnalyser system (Agilent Technologies, Palo Alto, CA, USA). Total RNA (1 μg) from each sample was used to generate a complementary DNA (cDNA) template using the iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA, USA). The total reaction volume was 20 μl. The reaction was incubated in a Tetrad thermocycler (MJ Research, Waltham, MA, USA) as follows: 5 min at 25°C, 30 min at 42°C, 5 min at 85°C, hold at 4°C. cDNA products were diluted 1:10 in diethylpyrocarbonate-treated water. Controls were also performed with no RNA template (no template) and omitting the RT enzyme (no RT).
The PCR primer pairs were designed based on previously published sequences (GenBank) using the Oligo 6 primer design software (Molecular Biology Insights). The PCR primer pair sequences, annealing temperatures, accession numbers and PCR product lengths are outlined in Table S1 of the supplementary material (available online at http://www.journals.cambridge.org/bjn.org). PCR was performed on cDNA using Taq DNA Polymerase obtained from Invitrogen (Carlsbad, CA, USA) and using previously described conditions(Reference Hontecillas, Wannemeulher and Zimmerman8, Reference Bassaganya-Riera, Pogranichniy and Jobgen35), and each gene amplicon was purified using the MinElute PCR Purification kit (Qiagen). The purified amplicon for each gene was quantified on an agarose gel and also with the GeneQuant Pro spectrophotometer (Amersham Biosciences, Piscataway, NJ, USA). These purified amplicons were further used to optimise the real-time PCR conditions and to generate the standard curves in the real-time PCR assay. Primer concentrations and annealing temperatures were optimised for the iCycler iQ System (Bio-Rad) for each set of primers using the system's gradient protocol. PCR efficiencies were maintained at 100 % for each primer set during optimisation and also during the real-time PCR of sample cDNA.
Statistical analyses
ANOVA was used to determine the statistical significance of the model: main effects of diet, genotype, time, two-way and three-way interactions when time was a factor. ANOVA was performed using the general linear model procedure of Statistical Analysis Software (SAS Institute, Inc., Cary, NC, USA) as described previously(Reference Bassaganya-Riera, Guri and Noble36). Data were analysed as factorial arrangements of treatments. The statistical model was
where μ is the general mean, genotypei is the main effect of the ith level of the genotypic effect (expression of PPARγ by the immune and epithelial cells), dietj is the main effect of the jth level of the dietary effect (PUA v. control), (genotype × diet)ij is the interaction effect between genotype and diet, and error A is the random error. When the model was significant, the analysis was followed by Scheffe's multiple comparison method. Data are expressed as means with their standard errors of the mean. For analysing the results of the disease activity index over time, we used a three-factor repeated-measures ANOVA. For this analysis, in addition to the main effects of diet and genotype and the two-way interaction between diet and genotype (as shown earlier), the model included the main effect of time, the diet × time, genotype × time interactions and the three-factor interaction (diet × genotype × time). Statistical significance was assessed at a probability value (P < 0·05).
Results
Disease activity indices
No effect of PUA was observed in disease activity indices of wild-type mice (Fig. 1(A)). PUA prevented experimental IBD in IL-10− / − mice (Fig. 1(B)). Even though there were some favourable numerical differences, PUA did not cure IBD in mice that received it after having developed severe clinical signs such as rectal prolapses (Fig. 1(C)). The deficiency of PPARγ in immune and epithelial cells in PPARγ fl/fl; MMTV-Cre+; IL-10 DK mice abrogated the beneficial effect of PUA in experimental IBD (Fig. 1(D)) even when PUA was administered preventively. Even though we also attempted to generate a line of PPARδ; IL-10 DK, we were unable to produce this line due to embryonic mortality associated with this genotype. Thus, we evaluated the role of PPARδ in PUA-mediated protection from IBD using a model of DSS colitis.

Fig. 1 Effect of punicic acid (PUA) on disease activity indices during 42 d. (A) C57BL/6J wild-type mice fed either a control (–■–) diet or a diet supplemented with PUA (1 g/100 g; –⋄–). (B) IL-10-deficient mice with no signs of disease before day 0 of the study (IL-10− / − P). (C) IL-10− / − mice with severe inflammatory bowel disease on day 0 of the study (IL-10− / − T). (D) PPARγ fl/fl; MMTV-Cre+; IL-10− / − double knockout mice. * Mean values were significantly different between the treatments (P < 0·05, n 10).
Intestinal inflammatory lesions
The architecture of colons recovered from IL-10− / − P mice administered PUA resembled those of healthy wild-type mice. Specifically, PUA significantly decreased the histological scores, including lymphoplasmacytic infiltration and enlargement of the colonic mucosa of IL-10− / − P, but not in IL-10− / − T, mice or in PPARγ fl/fl; MMTV-Cre+; IL-10 DK mice (Fig. 2).

Fig. 2 Effect of punicic acid (PUA) on microscopic lesions observed following 42 d of dietary supplementation. Representative photomicrographs of colons of (A and D) wild-type, (WT; B and E) IL-10− / − P and (C and F) IL-10− / − T mice fed a control diet (A–C) or a PUA-supplemented diet (D–F) for 42 d. The intestinal specimens were excised, stored in formalin, sectioned and stained with haematoxylin and eosin. Original magnification at 40 × . (G) Wild-type, IL-10− / − P and IL-10− / − T and PPARγ fl/fl; MMTV-Cre+; IL-10− / − double knockout mice were fed either a control diet (■) or a PUA-supplemented diet (□) for 42 d. Colonic histological lesions were scored by blinded observers based on size and morphology (0–4) as described in the Materials and Methods section. a,b Mean values with unlike letters were significantly different (P < 0·05, n 10).
Quantification of colonic gene expression
Quantitative real-time RT-PCR analyses demonstrated that colonic PPARδ was significantly up-regulated, and the expression of PPARδ-responsive gene angiopoietin-like 4 was numerically increased by PUA (Fig. 3(A) and (B)). No differences were observed in the colonic expression of PPARα, γ or their responsive genes CD36, FABP4 and stearoyl coenzyme A desaturase 1 (data not shown). PUA suppressed the colonic expression of both TNF-α and MCP-1 (Fig. 3(C) and (D)). PUA significantly up-regulated the colonic expression of keratinocyte growth factor in comparison with control diet-fed mice (0·01 v. 0·005, P < 0·02). Kerationocyte growth factor is a growth factor associated with epithelial wound healing.

Fig. 3 Quantification of mRNA expression of (A) PPARδ, (B) angiopoietin-like 4, (C) TNF-α and (D) monocyte chemoattractant protein 1 in colons of IL-10− / − mice fed a control diet or a punicic acid (PUA)-supplemented (1 g/100 g) diet using real-time RT-PCR. Colonic samples were collected from IL-10− / − mice fed PUA preventively. Values are means, with standard errors represented by vertical bars (ten mice per group). * Mean values were significantly different (P < 0·05). cDNA, complementary DNA.
Effect of cell-specific deficiency of PPARγ and δ on the ability of punicic acid to prevent or ameliorate dextran sodium sulphate colitis
PUA protected wild-type mice from experimental IBD, but its beneficial effects in disease activity and colonic lesions were abrogated in PPARδ null mice (Fig. 4) and significantly impaired in IEC-specific (Villin-Cre) (Fig. 5) and macrophage-specific (Lysozyme M-Cre) PPARγ null mice (Fig. 6), suggesting that PPARγ and δ in immune and epithelial cells are required for PUA-mediated protection from experimental IBD. The highest disease activity was observed in Lysozyme M-Cre mice, regardless of the diet (Fig. 6), indicating that the deficiency of PPARγ in macrophages is a particularly important contributor to the immunopathogenesis of IBD, as shown previously(Reference Shah, Morimura and Gonzalez29). Flow cytometric analyses of T-cell subsets in blood and mesenteric lymph nodes (MLN) demonstrated that PUA increased the percentages of regulatory T-cells in the blood of wild-type mice but not in mice lacking PPARγ or δ in immune or epithelial cells (Fig. 7).

Fig. 4 Effect of dietary punicic acid (PUA) supplementation on experimental inflammatory bowel disease during a 7 d challenge with dextran sodium sulphate. (A) Disease activity indices, (B) gross lesions in C57BL/6J wild-type (WT) and PPARδ null mice fed either a control or a PUA-supplemented diet. Values are means, with standard errors represented by vertical bars. a,b,c,d Mean values with unlike letters were significantly different (P < 0·05, n 10). – – –, WT control; – - –, WT PUA; ——, PPARδ control; ……, PPARδ PUA.

Fig. 5 Effect of dietary punicic acid (PUA) supplementation on experimental inflammatory bowel disease during a 7 d challenge with dextran sodium sulphate. (A) Disease activity indices, (B) gross lesions in Villin-Cre–C57BL/6J wild-type (WT) and intestinal epithelial cell-specific PPARγ null (Villin-Cre+) mice fed either a control diet or a PUA-supplemented diet. Values are means, with standard errors represented by vertical bars. a,b,c,d Mean values with unlike letters were significantly different (P < 0·05, n 10). – – –, WT control; – - –, WT PUA; ——, VillinCre+control; ……, VillinCre+PUA.

Fig. 6 Effect of dietary punicic acid (PUA) supplementation on experimental inflammatory bowel disease during a 7 d challenge with dextran sodium sulphate. (A) Disease activity indices, (B) gross lesions in Lysozyme M-Cre–C57BL/6J wild-type (WT) and macrophage-specific PPARγ null (Lysozyme M-Cre+) mice fed either a control diet or a PUA-supplemented diet. Values are means, with standard errors represented by vertical bars. a,b,c Mean values with unlike letters were significantly different (P < 0·05, n 10). – – –, WT control; – - –, WT PUA; ——, Lysozyme M Cre+control; ……, Lysozyme M Cre+PUA.

Fig. 7 Dietary punicic acid modulates the percentages of regulatory T-cells in the peripheral blood of C57BL/6J wild-type (WT) mice but not in PPARδ null, macrophage-specific PPARγ null (Lysozyme M (LysM)-Cre+), intestinal epithelial cell-specific PPARγ null (Villin-Cre+ (VC+)) mice with dextran sodium sulphate (DSS) colitis. Values are means, with standard errors represented by vertical bars. a,b,c Mean values with unlike letters were significantly different among the treatments (P < 0·05, n 10). PPARdKO, PPAR double knockout; WT no DSS, WT without DSS.
Discussion
Nutritional influences can target the main components of mucosal homeostasis during IBD and contribute to either attenuating or accentuating the onset of disease(Reference Cantorna, Munsick and Bemiss37, Reference Geerling, Badart-Smook and Stockbrugger38). Both fatty acid composition of the diet and total amount of dietary fat(Reference Calder39–Reference Liu, Duysen and Yaktine41) define the variables of lipid nutrition that influence health and disease. For instance, mixed results are available on the modulation of intestinal inflammation by n-3 PUFA, although CLA has shown anti-inflammatory efficacy more consistently, primarily by targeting PPAR(Reference Bassaganya-Riera and Hontecillas42). At the molecular level, PPAR represent important targets for the actions of dietary lipid(Reference Jump and Clarke43) and major contributors to the maintenance of intestinal homeostasis. In this regard, PPARγ gene therapy enhances PPARγ mRNA expression, resulting in dramatic therapeutic benefits in the DSS colitis model(Reference Katayama, Wada and Nakajima2). CLA induced colonic PPARγ expression and provided protection against the disease in a pig model of bacterial-induced colitis(Reference Hontecillas, Wannemeulher and Zimmerman8), as well as in mouse and pig models of DSS colitis(Reference Bassaganya-Riera, Reynolds and Martino-Catt3, Reference Bassaganya-Riera and Hontecillas9). The present study investigates the possibility of a PPAR-dependent mechanism underlying the anti-inflammatory efficacy of PUA against experimental IBD.
PPARγ and δ are recognised as central inhibitors of intestinal inflammation in DSS colitis(Reference Su, Wen and Bailey44–Reference Nakajima, Wada and Katayama47). In addition, activation of PPARγ by rosiglitazone ameliorated spontaneous pan-enteritis caused by the deficiency of IL-10(Reference Lytle, Tod and Vo4). In the present study, we provide evidence that preventive administration of PUA ameliorated IBD in two mouse models of IBD. However, PUA was not effective in IL-10− / − mice with established severe inflammatory lesions (i.e. rectal prolapses) and PPARγ; IL-10 DK mice. The latter finding suggests that the anti-inflammatory efficacy of PUA depends on the expression of functional PPARγ in immune and epithelial cells. Interestingly, colonic expression of PPARδ and its responsive gene angiopoietin-like 4 was up-regulated in IL-10− / − mice that received PUA preventively. These in vivo findings were in line with increased PPARδ reporter activity induced by PUA in vitro in IEC and macrophages. As CLA(Reference Bassaganya-Riera and Hontecillas9), PUA up-regulated colonic kerationocyte growth factor levels in the present study. Since PPARδ plays an important role in re-epithelialisation in mouse epidermis(Reference Michalik, Desvergne and Tan48), the up-regulated colonic keratinocyte growth factor may be indicative of a PPARδ-mediated re-epithelialisation of the gut mucosa.
In contrast to PPARδ and its responsive genes, colonic levels of PPARγ, α and their responsive genes remained unchanged. Nonetheless, since PUA transactivates PPARγ in 3T3-L1 pre-adipocytes(Reference Hontecillas, O'Shea and Einerhand11) and given the abrogation of the effect of PUA that we observed in PPARγ; IL-10 DK mice, this isoform was also investigated as a putative target for PUA. PPARγ suppresses inflammation by antagonising NF-κB, STAT and AP-1(Reference Ricote, Li and Willson49), favouring the nucleocytoplasmic shuttling of the activated p65 subunit of NF-κB(Reference Kelly, Campbell and King50), and SUMOylation of PPARγ results in a stable repressed state of NF-κB(Reference Pascual, Fong and Ogawa51). Thus, the down-regulation of TNF-α in colons of PUA-fed mice and M1 macrophages treated with PUA is consistent with the PPARγ-dependent anti-inflammatory effects of this compound.
The selective PPARγ agonist rosiglitazone suppressed colonic inflammation even when PPARγ was deleted from colonic epithelial cells, suggesting either an epithelial PPARγ-independent effect or a possible role for macrophages as a cellular target(Reference Adachi, Kurotani and Morimura26). In addition, activation of PPARγ and δ has been shown to suppress M1 classically activated or pro-inflammatory macrophage activation and favour M2 alternatively activated or anti-inflammatory macrophage differentiation(Reference Kang, Reilly and Karabacak52–Reference Odegaard, Ricardo-Gonzalez and Red Eagle54). Moreover, PPARγ and δ have been shown to exert overlapping anti-inflammatory effects in lipopolysaccharide-stimulated macrophages(Reference Welch, Ricote and Akiyama55). Based on this background, to further characterise the putative roles of PPARγ and δ as targets for PUA, we determined whether the deletion of these genes impaired or abrogated its ability to ameliorate experimental IBD. Our data demonstrate that both PPARγ and δ are required for PUA-mediated protection from DSS colitis. Additionally, PPARγ was also required for PUA-mediated protection from IL-10-induced pan-enteritis since the preventive effect of PUA was abrogated in PPARγ; IL-10 DK mice. However, we could not test the role of PPARδ in spontaneous pan-enteritis in IL-10 knockout mice since PPARδ; IL-10 DK mice did not survive beyond the embryonic stages.
At the cellular level, the deletion of PPARγ in macrophages completely abrogated the beneficial effect of PUA, whereas its deletion in IEC or the whole-body deletion of PPARδ impaired, but did not completely abrogate, the anti-inflammatory activity of PUA in the gut. Together, these data indicate that PUA ameliorates experimental IBD by down-modulating inflammation in mucosal immune and epithelial cells through PPARγ- and δ-dependent mechanisms. In support of this assertion, we provide in vitro evidence demonstrating that PUA treatment suppressed the TNF-α- and MCP-1-producing abilities of wild-type M1 classically activated macrophages, but it failed to exert these suppressive effects in PPARγ or δ null macrophages. Furthermore, PUA intake increased the peripheral blood regulatory T-cell compartment in wild-type mice but not in PPARγ or δ null mouse strains. These findings are in line with a PPARγ-dependent up-regulation of Foxp3 in regulatory T-cells treated with PUA (data not shown). Of note, regulatory T-cells mediate protection from experimental colitis through PPARγ-dependent mechanisms(Reference Wohlfert, Nichols and Nevius28, Reference Hontecillas and Bassaganya-Riera34). Since colonic PPARγ was required for some of the anti-inflammatory effects of PUA in vivo, but it did not activate PPARγ reporter activity directly, further studies are required to determine whether IEC and/or immune cells produced endogenous PPARγ agonists in response to PUA-mediated activation of PPARδ. In conclusion, PUA prevented experimental IBD through a mechanism requiring adequate expression of PPARγ and δ in immune cells and IEC in the colonic mucosa.
Acknowledgements
The present study was supported by funds from Lipid Nutrition BV. J. B.-R., R. H., Z. E. J., M. O'. S. and A. W. C. E. designed the experiments, interpreted the results, wrote the manuscript and managed the project. M. D., M. C., C. V. and A. C. conducted the animal studies. M. C. and C. V. conducted the in vitro studies. J. B.-R. filed a patent related to PUA. R. H., Z. E. J., M. O'. S., A. W. C. E., M. D., M. C., C. V. and A. C. declare that they have no conflict of interests.








