


Interest in tardive dyskinesia is reviving on the basis of recent evidence of prevalence with newer antipsychotic drugs similar to that reported with older ones (Carbon Reference Carbon, Hsieh and Kane2017) and new treatments receiving regulatory approval in the USA, a process likely to extend internationally. It is therefore appropriate to revive tardive dyskinesia in terms of clinical awareness and therapeutic options. The author's previous article addressed the first of these aims (Owens Reference Owens2018); the present addresses the second.
Aetiology
The most powerful method for devising targeted treatments in medicine is to test candidates derived from robust theories of disease aetiology. As in virtually all areas of psychopharmacology, treatment of tardive dyskinesia has been – and remains – profoundly constrained by the limitations of available theories of causation (Box 1). We do not understand the mechanism(s) underlying this common disorder, a major factor in large part because of the lack of satisfactory laboratory – especially animal – models (Waddington Reference Waddington1990).
BOX 1 The major pathophysiological theories of tardive dyskinesia aetiology
Postsynaptic D2 receptor supersensitivity
Upregulation of dopamine D2 receptors in response to chronic blockade, creating dopamine–acetylcholine imbalance
Simplest theory but riddled with problems
Presynaptic dopamine/noradrenaline hyperactivity
Pioneering shift in emphasis to presynaptic mechanisms
Well-argued but on outmoded biological parameters
Never fully exploited
Cholinergic interneuron burnout
Chronic dopamine blockade, resulting in loss of inhibition on large cholinergic interneurons, with consequent ‘burnout’
Postulates similar proximate pathology as above but allows for consideration of other transmitter systems
Excitatory/oxidative stress (neurodegeneration)
Increased dopamine turnover, resulting in loss of function of large, inhibitory GABAergic aspiny interneurons, (and/or medium spiny neurons), possibly from membrane damage
Some experimental support
Falls on failure of antioxidants as effective treatment
Disordered synaptic plasticity
Upregulated dopamine receptors on medium spiny (and other) neurons adversely influence glutamatergically mediated synaptic plasticity, unbalancing flow between ‘direct’ and ‘indirect’ striatal pathways, compounded by similar problems at cortical dopamine–glutamate sites
Innovative – most persuasive in relation to stereotyped oromandibular disorder and dystonia
Nonetheless, since tardive dyskinesia is associated with compounds – antipsychotics and others – that block central dopamine transmission, this has understandably been the favoured focus.
Dopamine
The first and most persistent hypothesis of tardive dyskinesia aetiology relates to postsynaptic dopamine (D2) receptor supersensitivity. This was proposed independently in 1970 by neurologist Harold Klawans and Nobel Laureate pharmacologist Arvid Carlsson, by analogy with the outwardly similar abnormalities evident in people taking long-term L-dopa for parkinson's disease. The idea is that chronic antagonist blockade results in increased postsynaptic dopamine receptor numbers (and/or sensitivity of unblocked receptors), resulting, in line with Cannon's law of denervation, in a supersensitised motor system. In functional terms, this would place tardive dyskinesia as the pathophysiological ‘opposite’ of parkinsonism – domination of striatal dopaminergic activity rather than its increasing subtraction (Fig. 1). The promoting factor may be dopamine excess but the symptom mediator is cholinergic insufficiency.
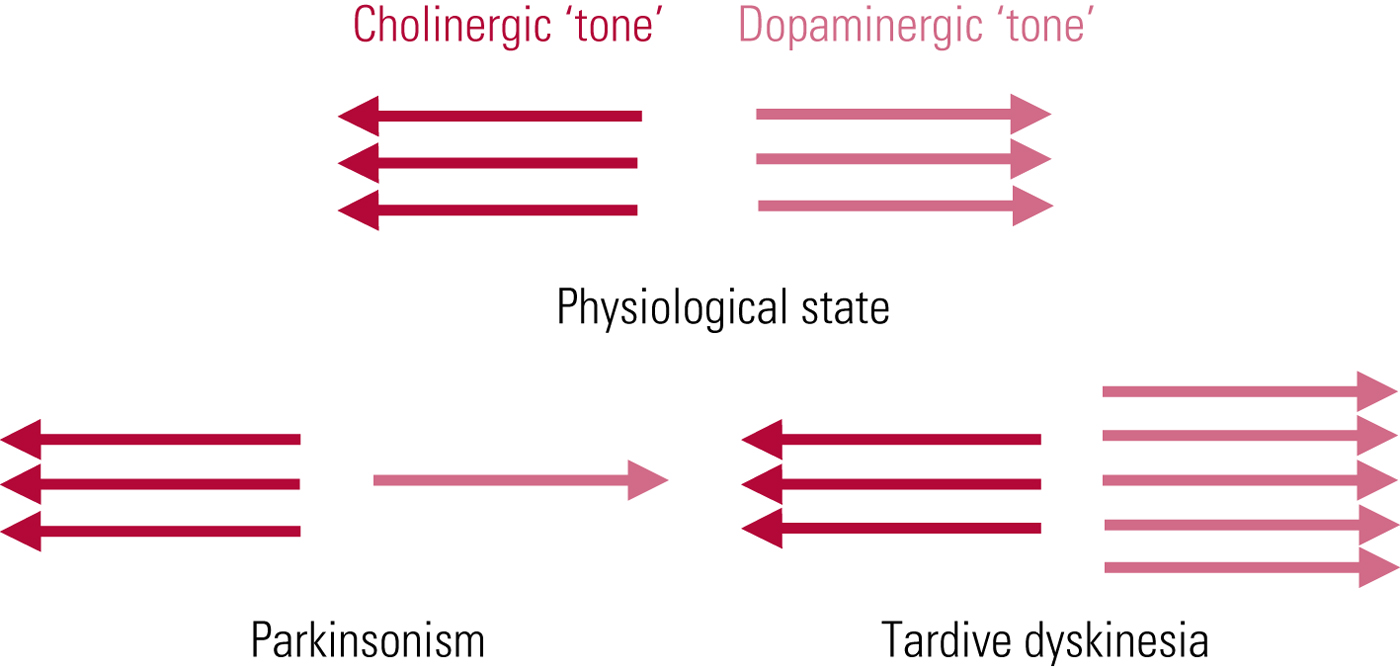
FIG 1 Tardive dyskinesia: schematic representation of functional implications of the postsynaptic receptor supersensitivity hypothesis. From Owens (Reference Owens2014).
The value of the theory is that it explains the phenomenon of ‘suppression’, where even quite marked degrees of tardive dyskinesia can be, at least temporarily, ameliorated or abolished by antipsychotic dose escalation. However, cross-sectional comparisons of tardive dyskinesia and L-dopa-induced dyskinesias reveal similarity, not identity (Karson Reference Karson, Jeste and LeWitt1983), and this theory raises more questions than it answers (Owens Reference Owens2014). At best, it should be seen as providing limited insight.
Beyond dopamine
Which means coming up with an alternative! Although several have been proposed, thus far none provides compelling understanding.
1 Presynaptic dopaminergic/noradrenergic hyperactivity hypothesis (Jeste Reference Jeste and Wyatt1981): an early and neglected challenge to conventional wisdom came when emphasis was first shifted to presynaptic mechanisms, with the proposal that presynaptic dopaminergic overactivity and noradrenergic hyperactivity were together necessary for at least some types of tardive dyskinesia. Beta-blockers provide a ready test of this hypothesis but have been surprisingly neglected as potential therapeutic options, although interest is reviving (Hatcher-Martin Reference Hatcher-Martin, Armstrong and Scorr2016). This is a theory whose implications are now topical but remain to be exploited.
2 Cholinergic interneuron burnout hypothesis (Miller Reference Miller and Chouinard1993): acetylcholine plays a crucial role in striatal function, the striatum being the source of the highest levels of acetylcholine markers (transmitter plus synthesising/metabolising enzymes) in the brain. Most of this acetylcholine is associated with cholinergic interneurons, although these account for only 1–2% of striatal cell numbers, a feat achieved through extreme arborisation. Under physiological conditions, cholinergic interneurons tonically discharge, a process modulated by dopaminergic inhibition. The proposal is that with long-term dopamine blockade, cholinergic interneurons, in effect, burn out from loss of inhibitory control. This hypothesis was an interesting departure from the sterility of supersensitivity, not least because it opens up for consideration a wide range of other transmitter systems that themselves interact with cholinergic interneurons, such as GABAergic (Fibiger Reference Fibiger and Lloyd1984) and glutamatergic, both ionotropic and metabotropic (Lim Reference Lim, Kang and McGehee2014). However, as with supersensitivity, confirmation is lacking.
3 Excitatory/oxidative stress hypothesis (Lohr Reference Lohr1991): the notion that disease can be mediated via ultra-brief, highly reactive chemical moieties is well-rooted in pathology. In relation to tardive dyskinesia, the proposal is that increased presynaptic dopamine activity (turnover), initially identified with postsynaptic antagonism, creates damaging dopamine quinones, and possibly hydrogen peroxide, through the action of monoamine oxidase. A further suggestion is that damage emanates from deficient production of free-radical defences, such as superoxidase dismutase (Cadet Reference Cadet, Lohr and Jeste1986), and increased lipid peroxidation, which result in membrane damage/dysfunction and possible cell death (Elkashef Reference Elkashef and Wyatt1999). Whatever the mechanism, the consequence is degeneration of dopamine-receptor-rich, GABAergic striatal interneurons, themselves normally inhibitory. A degree of experimental evidence supports this hypothesis in, for example, depositions of heavy metals and elevated cerebrospinal fluid (CSF) lipid peroxidase parameters but, again, it has not matured beyond a theoretical proposal. A major impediment has been the generally disappointing results of antioxidants as treatments.
4 Synaptic plasticity hypothesis (Teo Reference Teo, Edwards and Bhatia2012): this taps into a topical field in neuroscience and, according to its proposers, could provide the missing link between supersensitivity and interneuron dysfunction. It sets out a number of possibilities, one of which is that D2 hypersensitisation (of striatal medium spiny neurons and feedforward fast-spiking interneurons) affects the plasticity of glutamatergic interneuron synapses, resulting in disordered striato–thalamo–cortical output to the sensorimotor cortex, one consequence being reduced activity in cortical inhibitory circuits, with increasing excitability in executive motor areas. Such miscoding of motor programmes could explain persistence of disorder after removal of the causative antipsychotic. This elegant theory is in line with modern proposals on the pathophysiology of other hyperkinetic states, especially chronic dystonia (Calabresi Reference Calabresi, Pisani and Rothwell2016), and may have its greatest relevance to disorder of this type but remains another hypothesis without validation.
It is possible each of these broad brushes paints part of the picture but none, as yet, allows us to make that crucial leap to innovative therapy.
Management and treatment
Primary prevention
In the absence of treatments with authentic antidyskinetic actions, the management of tardive dyskinesia remains firmly rooted in primary prevention – limiting indications, limiting doses and avoiding scenarios of heightened risk (Box 2).
BOX 2 Principles of primary prevention
Limiting indications
Psychiatrists should understand the nature of drug licensing procedures and the marketing strategies they are designed to foster.
Limiting doses
Wide pharmacokinetic diversity means that individualised tailored dosing must be derived empirically, with licensed/approved doses only a rough guide.
The mantra is ‘start low, rise slow’, allowing adequate time for biological systems to adapt before implementing incremental planning.
Avoiding scenarios of heightened risk
This is most difficult in practice as ‘risk’ cannot always be identified in advance of tardive dyskinesia emerging.
Remember predisposing factors (Owens Reference Owens2018) and:
• minimise risk of ‘acute-end’ EPS (‘start low, rise slow’)
• be wary of high-potency drugs in older patients
• avoid drug-free intervals of longer than 1 month (needs particular consideration in those with established relapsing–remitting illnesses)
• monitor closely in those with clear organic pointers.
Antipsychotic prescribing has increased fourfold in the past two decades, mainly from a rise in the use of newer compounds for an expanding catalogue of licensed indications (Verdoux Reference Verdoux, Tournier and Begaud2010). It is essential to appreciate that ‘licensing’ is a commercial imperative, where companies (‘marketing authorisation holders’) are required to present efficacy and safety data to support product promotion. It does not mean that licensed products are the only drugs for a particular job – nor that a particular ‘job’ requires a product at all! Antipsychotics, as the name implies, should be restricted to ‘psychotic’ indications and not used for management of non-specific problems such as insomnia or disruptive behaviours, unless in extremis and only when all other pharmacological and psychological routes have been exhausted. It was only in the 1970s that the original inference of ‘psychosis’ (‘severity’) was supplanted by a concept based on the presence of particular symptoms (hallucinations, delusions, thought disorder and sometimes affective incongruity). In clinical practice, ‘antipsychotics’ would be appropriately indicated in mania/hypomania, traditionally considered ‘psychotic’ disorders.
Minimum effective dosing is the key feature of antipsychotic use but, because of pharmacokinetic diversity, is difficult to standardise across patients. The physician's skill is in tailoring therapy to individual need by choice of compound and dose, both initial and incremental, a key element of which is adopting realistic timescales to allow for adaptation of biological systems. Pragmatic trials have consistently failed to support the view that newer antipsychotics have an inherently reduced liability to promote extrapyramidal side-effects (EPS) (Lieberman Reference Lieberman, Stroup and McEvoy2005; Jones Reference Jones, Barnes and Davies2006; Fischer-Barnicol Reference Fischer-Barnicol, Lanquillon and Haen2008), leading to the inevitable conclusion that their advantage lies in licensed doses, pharmacokinetically derived, being lower than the empirically derived doses traditionally utilised with older drugs (Owens Reference Owens2014). One might postulate that the comparability of tardive dyskinesia rates that has emerged with longer-term use of newer compounds relates to the fact that, in practice, doses of these tended to rise post-launch (Citrome Reference Citrome, Jaffe and Levine2005, Reference Citrome and Kantrowitz2009).
Risky prescribing cannot always be avoided but the prescriber's responsibility is to be aware of variable risk. For example, the ageing brain does not tolerate antipsychotics well, and utilising this class in older patients, especially with first exposure, needs careful thought.
Pharmacological treatment
Even with diligent care, individual predisposition dictates that some patients will still develop disorder but, as a result of multifarious and sometimes competing confounds in inadequately powered studies, the research literature on their treatment is of ‘poor’ or ‘very poor’ quality (Bergman Reference Bergman, Walker and Nikolakopoulou2017) and it is not possible to specify a definitive algorithm.
Assessing the efficacy of treatments for tardive dyskinesia is uniquely complex (Box 3). The condition itself comprises different types of disorder (Owens Reference Owens2018), which may be differentially affected by therapeutic interventions, and a large part of its clinical presentation is exquisitely sensitive to modifiers, both external and internal (environment, mental state, etc.). As a result, tardive dyskinesia is highly variable over time, even within the day. Coincidental medication can have an apparently favourable effect but for entirely non-specific reasons. There are issues surrounding the recording instrument chosen, how comprehensive the assessment is and how data are analysed. Importantly, there issues about how one militates against unconscious biases, which extends beyond just treatment allocation (see below: critique). Not least is power. Perhaps the ideal study is un-doable but in appraising those that do come to publication, it is important to be aware of these complexities.
BOX 3 Some points for consideration in the design/interpretation of therapeutic studies in tardive dyskinesia
1 Type of recording instrument
Multi-item (specific predefined movements/behaviours) versus global impression (all movements/behaviours in specified areas)
2 Sample homogeneity
What is the target disorder type (oromandibular/limb–truncal, with or without parkinsonism)?
3 Stability of routine therapies
Antipsychotics unchanged (for ? 3 months), and avoidance of other drugs, especially with sedative or stimulant actions (e.g. benzodiazepines, antimuscarinics) throughout the study period
4 Fixed assessment time/location
Morning/afternoon; familiar (ward) or unfamiliar (examination room) location
5 Coincidental psychiatric symptomatology
Variations in psychotic and non-psychotic features
6 Nature of placebo
Inactive versus active (to account for effect of non-target actions)
7 Adequate run-in
To allow participants to adapt to trial conditions
8 Adequate sample size
To power for a clinically meaningful effect
9 Adequate trial period
To ensure validity of effect
10 Method of analysis
Total scores versus means or other measures of change (e.g. single symptoms)
11 Masking (‘blinding’)
To treatment allocation and effects of contextual bias
(After Owens Reference Owens2014)
Antipsychotic dose reduction/cessation/switching
There is no persuasive evidence that intuitive approaches such as dose reduction, cessation or switching to another antipsychotic are effective in either management or treatment (Bergman Reference Bergman, Rathbone and Agarwal2018). Clinical emergence of tardive dyskinesia is the outward sign of major functional imbalance in motor systems that have probably been disrupted, but compensated, for a very long time but which are now settling into decompensated relationships that likely spell irreversibility. From the mental state perspective, cessation may not be an option, while dose reduction may actually promote exacerbation by addition of withdrawal-emergent effects that, in older adults at least, may not settle to pre-reduction levels.
This does not amount to advocacy for inaction – only realism. Early recognition is crucial and follows from regular monitoring, preferably with use of a standardised recording instrument, such as the Abnormal Involuntary Movement Scale (AIMS; Guy Reference Guy1976: pp. 534–7), which should be an integral part of follow-up paperwork. With early recognition, consideration of rationalisation is important, although if dose reduction is the favoured path, it should be undertaken very cautiously, in terms of both amounts and timescales (minimal decrements, weeks/months apart). Again, the purpose is to try to encourage any potential reversal of biological imbalances without radically overwhelming them.
Much of the limited work on switching seems predicated on acceptance of the newer antipsychotics as, pharmacologically, fundamentally different from what came before – hence their description by many as ‘atypical’. The clinical evidence (to say nothing of the comparable tardive dyskinesia prevalences that are the present focus) comprehensively discredits this proposal, although it remains stubbornly entrenched. The author would encourage clinicians not to consider antipsychotics in these terms, especially for the purposes of switching. A better approach might be to consider potency and receptor-binding profiles, with the aim of transferring – again, very slowly – to a compound of lower potency and broader receptor-binding profile. This is trying to substitute the cosh of high-potency antagonism with something gentler, while non-specific actions, such as sedation, may help minimise the expression of emergent disorder. Any efforts are nonetheless empirical, with limited expectations.
Antipsychotic dose increment
It has long been known that the most effective management of tardive dyskinesia is to increase antipsychotic dose (Jeste Reference Jeste, Lohr and Clark1988) (Table 1). This apparently contrary observation reflects the phenomenon of suppression, thought to result from further blockade of oversensitised dopamine receptors, or the related masking by emergent parkinsonism. It is a likely explanation for much ‘improvement’ in switching studies where the switch is to a more potent compound.
TABLE 1 Average rates of improvement in tardive dyskinesia in controlled treatment trials
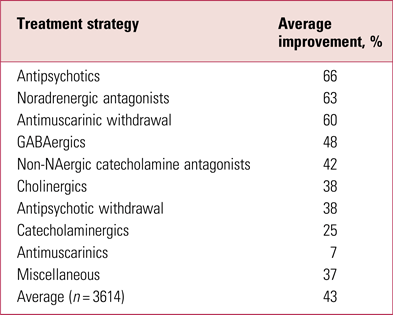
Source: Owens (Reference Owens2014); data from Jeste et al (Reference Jeste, Lohr and Clark1988).
Never popular, such strategies remain largely of theoretical interest and should only be considered in tertiary specialist centres.
Antioxidants
This approach, because of its simplicity and topicality, had great appeal in the 1990s. Several randomised controlled trials (RCTs), involving a total of just over 260 patients, suggested benefit from the free-radical scavenger alpha-tocopherol (vitamin E), although initial enthusiasm has not withstood systematic review (Soares-Weiser Reference Soares-Weiser, Maayan and McGrath2011; Bergman Reference Bergman, Walker and Nikolakopoulou2017). There is weak, unreplicated evidence that, if started early, vitamin E may slow progression (Adler Reference Adler, Edson and Lavori1998).
Other compounds advocated on the same theoretical basis but with equal lack of support include lipoic acid (Thaakur Reference Thaakur and Himabindhu2009) and the flavinoid rutin (Bishnoi Reference Bishnoi, Chopra and Kilkarni2007), although meta-analysis of small studies using adjunctive melatonin do suggest potential benefit (Sun Reference Sun, Zheng and Yang2017), and a single positive RCT supports Ginkgo biloba extract (Zhang Reference Zhang, Tan and Zhang2011). Clinical application of such approaches remains of the try-and-see variety.
After many years of neglect there is increasing evidence that free-radical scavengers do reduce development of vacuous chewing movements in rodents (Lister Reference Lister, Nobrega and Fletcher2014, Reference Lister, Andreazza and Navaid2017; Shi Reference Shi, Tan and Wang2016) long, if controversially, utilised as an animal model for tardive dyskinesia (Waddington Reference Waddington1990), so the ‘anti-oxidant story’ may not have run its course. Vitamin E (and other scavengers) is certainly safe and easily administered and may be worth considering at the first signs of persistent disorder, with the aim of minimising progression.
Clozapine
Clozapine is clearly associated with a reduced risk of extrapyramidal dysfunction in general, including tardive dyskinesia, although the magnitude of this benefit remains unclear (Owens Reference Owens2014). It would nonetheless be reasonable to assume that it may also offer benefit to those who have already developed disorder. Evidence is, however, poor (Hazari Reference Hazari, Kate and Grover2013) and use for a tardive dyskinesia indication must again be largely empirical.
Clozapine's value might best lie with tardive dystonias: although some suggest improvements may not continue after the first 3 months, others suggest that perseverance for up to 3 years is justified (Lieberman Reference Lieberman, Salz and Johns1991; Louza Reference Louza and Bassitt2005). The drug's strong antimuscarinic actions could be relevant to efficacy, although reports of withdrawal-emergent disorder following reduction/cessation (Ahmed Reference Ahmed, Chengappa and Naidu1998) also suggest a suppressant action or non-specific sedation.
Clozapine's benefits may represent active ‘treatment’ but may stem simply from maintenance of mental state stability while neurological signs settle spontaneously.
GABAergics
In the 1970s, demonstration of decreased dopamine transmission following augmentation of GABAergic mechanisms promoted gamma-aminobutyric acid (GABA) receptor agonism as a therapeutic strategy for tardive dyskinesia. An early supportive observation was the beneficial effect of benzodiazepines, whose action is based on GABA facilitation. However, benzodiazepines also have powerful sedative actions, which could favourably modify the expression of tardive dyskinesia, thereby aiding ‘management’ without necessarily affecting ‘treatment’. This is supported by the observation that benefits from clonazepam tend to reduce over several months, suggesting habituation to the drug (Thaker Reference Thaker, Nguyen and Strauss1990).
GABA facilitation approaches have been impeded by lack of probes with acceptable tolerability, and baclofen, gamma-vinyl-GABA, progabide, muscimol and valproate have been investigated only in small studies of poor quality that provide no evidence for therapeutic recommendations (Alabed Reference Alabed, Latifeh and Mohammad2011).
Cholinergics
The dopamine supersensitivity and cholinergic burnout hypotheses both implicate cholinergic mechanisms in symptom promotion, the one by their being overwhelmed, the other by their loss. There have, of course, long been tools with which to test this, and the older retrospective literature did report associations between antimuscarinic use and tardive dyskinesia risk. This was not supported by subsequent, largely prospective work, and was never disentangled from the issue that those on antimuscarinics were those most likely to have had ‘acute-end’ EPS, which may represent the genuine predisposition (Owens Reference Owens2014). The conclusion was that, although antimuscarinics may exacerbate tardive dyskinesia, especially choreiform-type disorder, it is unlikely that they are themselves causative agents (Gerlach Reference Gerlach1981).
Strategies to enhance striatal cholinergic transmission by boosting dietary precursors of acetylcholine, such as choline or lecithin, were widely pursued, but data are inadequate to form conclusions and, because of tolerability problems, the field is unlikely to develop further. However, long and safe use of acetylcholinesterase inhibitors, for which there is some evidence of benefit (Caroff Reference Caroff, Walker and Campbell2007), may point to an oversight in research focus (Tammenmaa-Aho Reference Tammenmaa-Aho, Asher and Soares-Weiser2018).
Cholinergic modification may, however, be relevant in the management of predominant or exclusive dystonia. Very high antimuscarinic doses (>100 mg/day trihexyphenidyl) were, and occasionally still are, recommended for idiopathic dystonia (Jankovic Reference Jankovic2009), although because of their poor tolerability. The author has not generally had positive results using them with psychiatric patients. Such tactics should be left to movement disorder specialists.
Presynaptic depleters
The surge of publications on tardive dyskinesia in recent years has been driven by the fact that two variants on this theme (valbenazine and deutetrabenazine: see below) have received US licenses. It is important to see these developments in context and to evaluate what is on offer hard-headedly.
Relevant neurophysiology
Recycled neurotransmitters are taken up across the presynaptic membrane by specific protein transporters, and stored, as is newly synthesised transmitter, in vesicles, which are mainly formed by budding from endoplasmic reticulum (Fig. 2). Vesicular monoamine transporter 2 (VMAT2) is a glycoprotein with 12 transmembrane domains that acts to facilitate the uptake of cytosolic transmitter into these storage vesicles to await use/reuse.Footnote a This is an active, energy-dependent process involving one of the widely expressed adenosine triphosphatase (ATPase) enzymes, vacuolar-type H-ATPase (V-ATPase) which, by pumping protons into newly formed vesicles, produces a conformational change in the transporter, allowing importation of transmitter. Excess transmitter left in the cytosol is broken down by monoamine oxidase. VMAT2 is non-selective and deals with re-storage of serotonin and histamine as well as the catecholamines, dopamine and noradrenaline.
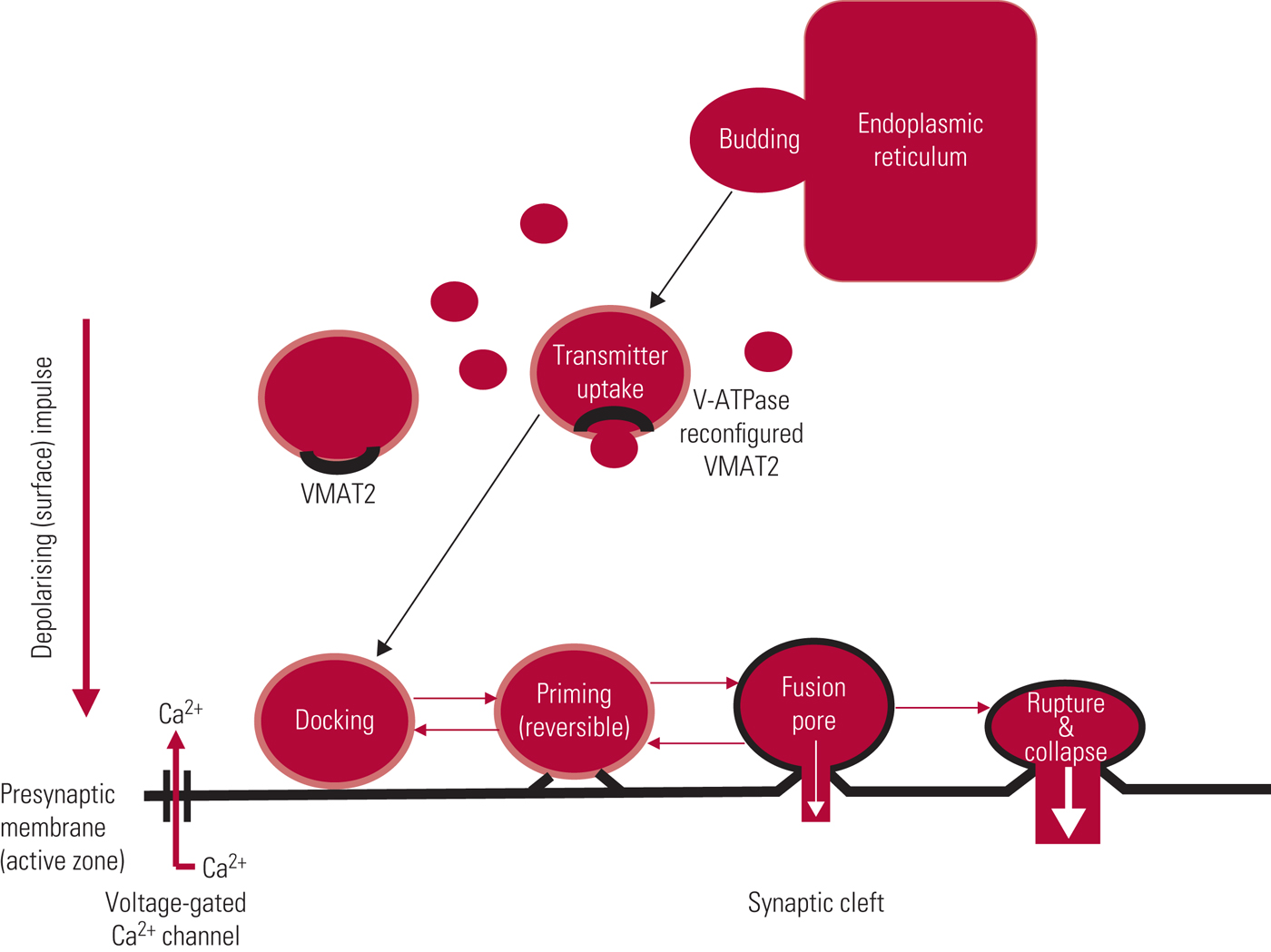
FIG 2 Storage vesicles: life-cycle and pharmacological modification. After Arbuthnott & and Garcia-Munoz (Reference Arbuthnott, Garcia-Munoz, Johnstone, Owens and Lawrie2010).
Vesicles are docked to the internal presynaptic membrane, most commonly by synchronous fusion, an immensely complex process involving recruitment of a series of soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) proteins and binding partners, where docking is precisely time-locked to terminal depolarisation (Li Reference Li and Kavalali2017). Transmitter release, by a process called exocytosis, follows influx of calcium, with docked vesicles releasing their content into the synaptic cleft either through generation of a fusion pore or by complete membrane fusion and rupture (Fig. 2). In most instances, the ‘empty’ vesicle is incorporated into the presynaptic membrane by collapse fusion. The presynaptic membrane also contains pits coated with the protein clathrin, which provide a source of new vesicle formation. Although incredibly complex, the vesicular cycle, from budding to fusion/collapse, can be completed within a minute (Li Reference Li and Kavalali2017).
The precision of synaptic messaging is such that variations in presynaptic release differentially affect downstream receptor activity almost instantaneously. In addition to theoretically addressing disease processes at origin, presynaptic depleters, mediating reduced transmitter release, represent a gentler, more subtle way of modulating synaptic events than ‘heavy-handed’ postsynaptic receptor antagonism.
Clinical pharmacology
The traditional approach: tetrabenazine
Extracts of Rauwolfia serpentina (Indian snakeroot) have been used in Ayurvedic medicine for centuries, and one of the first compounds to be refined from the plant, reserpine, was also one of the first drugs of the psychopharmacology age. Reserpine interacts with VMAT1 but also binds irreversibly to its own VMAT2 binding site. The drug fell from widespread psychiatric use because of poor tolerability. However, a synthetic analogue, tetrabenazine, which binds selectively to VMAT2 and reversibly to a separate, or possibly overlapping, site from reserpine (Grigoriadis Reference Grigoriadis, Smith and Hoare2017), was promoted as an antipsychotic in the UK from 1958 until its withdrawal in 1966 on a reputation of poor efficacy. It did, however, remain on the market in a number of European countries, including the UK, as a treatment for hyperkinetic movement disorders, including tardive dyskinesia. In the USA, approval was only obtained in 2008 and then for Huntington's disease alone – but with an unusual restriction. In addition to a black box warning in relation to depression and suicidality, the Food and Drug Administration (FDA) imposed the recommendation for CYP2D6 genotyping with higher doses. This might be seen as running ahead of the science and as contributing to an unnecessarily dark reputation. This difference in availability and perception between Europe and the USA is important in assessing how great a leap forward new compounds might be perceived as representing.
Although better tolerated than reserpine, tetrabenazine still has a prominent side-effect profile (Guay Reference Guay2010), some of which relates to the fact that several metabolites are active D1 and D2 antagonists (Grigoriadis Reference Grigoriadis, Smith and Hoare2017). Indeed, its tolerability profile is very suggestive of a mild, sedative antipsychotic (Table 2) – although presentation of adverse effects by patient report can be misleading: for example, ‘insomnia’ in a prominently sedative D2-modifying drug such as tetrabenazine is likely to reflect akathisia. In addition to patient-reported effects, tetrabenazine has a slight ability to prolong the QTc interval (according to the Summary of Product Characteristics by, on average, 8 ms) and caution is advised when co-prescribing potent CYP2D6 inhibitors. With a short half-life (approximately 2–3 h) and rapid and comprehensive brain penetrance but equally rapid egress, it requires dosing twice or, preferably, three times a day. Tetrabenazine has never been widely used in UK psychiatry, remaining a niche neurological product, and perhaps as a result is costly for a compound that has been around for so long.
TABLE 2 Adverse effectsa of tetrabenazine (>10%)
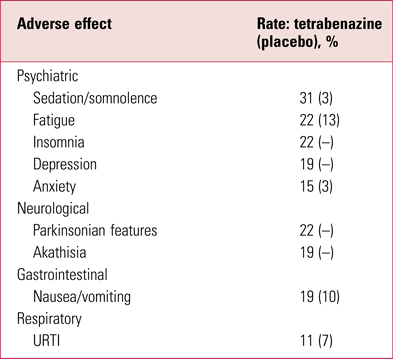
a Only those reported by >10% of patients are shown here.
The new approach: valbenazine and deutetrabenazine
Two approaches attempt to address the tolerability problems of tetrabenazine by modifying kinetics.
Tetrabenazine undergoes heavy first-pass metabolism, via mainly CYP2D6 (with 1A2 a minor pathway), resulting in bioavailability of less than 5%. From a complex range of outcomes, four discrete isomeric secondary alcohols are created, collectively referred to as dihydrotetrabenazine (Grigoriadis Reference Grigoriadis, Smith and Hoare2017). All four inhibit VMAT2 but one (R,R,R-HTBZ), the most potent inhibitor, has been esterified with the amino acid L-valine to produce valbenazine. The (R,R,R)-isomer demonstrates high specificity for the VMAT2 transporter while being virtually devoid of receptor interactions (Grigoriadis Reference Grigoriadis, Smith and Hoare2017).
Valbenazine acts as a prodrug which, following absorption, releases active HTBZ by hydrolysis to inhibit VMAT2. Amino acid esterification is increasingly implemented to improve the kinetic qualities of drugs of short half-life or with extensive first-pass, which require repeat dosing with resulting increases in peak-level side-effects (c.f. lisdexamfetamine).
The second method for modifying kinetics adopts a different, but again increasingly utilised, approach – deuteration. This involves replacing hydrogen atoms (in this case six), with deuterium, also known as heavy hydrogen (D or 2H), the resulting compound being referred to as deutetrabenazine. Deuteration slows a drug's metabolism without conveying other significant chemical changes.
Efficacy/tolerability of valbenazine and deutetrabenazine
Both valbenazine and deutetrabenazine have been shown to be effective in significantly reducing global tardive dyskinesia ratings using, as the primary outcome, total scores on the AIMS (O’Brien Reference O'Brien, Jiminez and Hauser2015; Anderson Reference Anderson, Stamler and David2017; Fernandez Reference Fernandez, Factor and Hauser2017; Hauser Reference Hauser, Factor and Marder2017), with 40% and 33% on the higher valbenazine (80 mg/day) and deutetrabenazine (36 mg/day) doses respectively achieving 50% or greater reduction in AIMS totals. Furthermore, both compounds are well tolerated (Table 3), although it is important to appreciate that adverse effects profiles, as presented in Tables 2 and 3, permit only relative, and not absolute, tolerability comparisons. Nonetheless, the likelihood of being helped or harmed (LHH), a ratio of the number needed to treat (NNT) to achieve a 50% reduction in AIMS totals over the number needed to harm (NNH) from discontinuation because of an adverse event, has been calculated at 15.2 for valbenazine and 27 for deutetrabenazine (Citrome Reference Citrome2017a,Reference Citromeb), projecting both drugs as highly favourable therapeutic agents.
TABLE 3 Adverse effects of valbenazine and deutetrabenazine
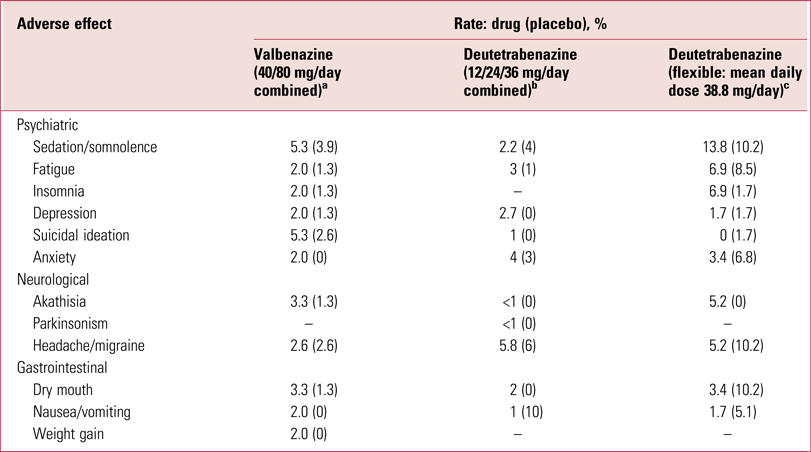
a. Data from Hauser et al (Reference Hauser, Factor and Marder2017).
b. Data from Anderson et al, Reference Anderson, Stamler and David2017).
c. Data from Fernandez et al (Reference Fernandez, Factor and Hauser2017).
Critique
Studies evaluating these compounds, which relate to short-term efficacy (valbenazine 6 weeks; deutetrabenazine 12 weeks), are of good quality (Davis Reference Davis, Miller and Kalsi2017), addressing a number of problems that so confounded past work, in particular the complex issue of masking (Box 3). This involves not just unawareness of treatment allocation but must take into account sequencing or expectancy bias (expectation of improvement as a study progresses) and the phenomenon of prior context (overflow of previous to current assessment), among other biases. Even though these issues were highlighted over two decades ago (Owens Reference Owens, Finlayson and Mercer1997) these are the first neuropharmacology arenas, to the author's knowledge, in which trial design included randomised masked video ratings. Both these programmes therefore give a major nod to point 11 in Box 3. By adopting a global impression recording instrument and presenting data as total scores, neither programme allows for judgements on what type of movement disorder – if any – is responding preferentially or in which body part it occurs, although further detail may emerge. Participants did partially fulfil the stability criterion in that they were required to have been on antidopaminergic medication for 3 months or more (>1 month in over-60s for deutetrabenazine) but regimes were only required to be stable for 30 days and additional medications, although discouraged, were permitted.
The profile of presynaptic depleters is likely to rise as they represent neglected drug targets for CNS disorders, especially in relation to schizophrenia, where their pharmacology fits well with the theoretical shift in the dopamine hypothesis from its original postsynaptic receptor focus to more recent emphasis on presynaptic mechanisms (Howes Reference Howes and Kapur2009). However, in fully evaluating these drugs – and justifying costs – we must consider research findings critically. In relation to tardive dyskinesia, a condition so readily modified by mental state and environment, an important confound relates to point 6 in Box 3 – the nature of the placebo. The question is the extent to which any benefits can be attributed to specific antidyskinetic pharmacological action(s), or result from non-specific factors such as sedation or suppression.
Tetrabenazine is quite markedly sedative (Table 2) but creating a pro-drug analogue or slowing kinetics appears to reduce this problem (Table 3). It is, however, difficult to assess the extent of this benefit, as direct head-to-head comparisons have not been undertaken. Data, as noted, only allow for relative not absolute conclusions, an important distinction (Rodrigues Reference Rodrigues, Duarte and Costa2017). Although well tolerated, both valbenazine and deutetrabenazine do have sufficient relevant actions to contribute to, if not account for, the relatively modest reductions in AIMS ratings reported.
Since deuteration favourably extends kinetics but does not fundamentally alter metabolism, one must, furthermore, assume that deutetrabenazine is associated with production of D2-antagonistic metabolites, opening another possible window on reduced AIMS ratings. Although parkinsonism was assessed in the valbenazine study using the much criticised Simpson–Angus Scale (SAS) (Owens Reference Owens2014), both deutetrabenazine studies used the motor subscales of the Unified Parkinson's Disease Rating Scale (UPDRS), a comprehensive and reliable instrument (Owens Reference Owens2014). Neither compounds were associated with significant increases in Parkinsonian ratings. This, however, does not mean that subthreshold increases in D2 antagonism are not relevant to the drug's action. Caution is supported by the fact that Anderson et al (Reference Anderson, Stamler and David2017) found that improvements on deutetrabenazine were greater in those not receiving a concomitant antipsychotic, compatible with the view that some benefit emanates from suppression.
In general, such comments might seem trifling if efficacy is striking. Presentation of trial results in terms of percentages achieving 50% reduction in signs certainly creates an impact but the actual drug-attributable reductions in both the valbenazine and deutetrabenazine studies were modest. On a scale with a possible total score of 21, reductions relative to placebo were, on average, 3.1 AIMS scale points for valbenazine 80 mg/day (1.8 for the 40 mg/day dose) (Hauser Reference Hauser, Factor and Marder2017) and less than 2 AIMS points for the two higher deutetrabenazine doses (Anderson Reference Anderson, Stamler and David2017; Fernandez Reference Fernandez, Factor and Hauser2017). Variance was, as would be expected, considerable. Although the video ratings showed statistically significant effects, the Clinical Global Impression ratings were not consistently significantly different (Fernandez Reference Fernandez, Factor and Hauser2017; Hauser Reference Hauser, Factor and Marder2017). This means that on-site clinicians dealing with the patients regularly could not necessarily detect the improvement trend. This is important, bearing in mind the biases, noted above, to which such assessors are prone.
Thus, there is reason to believe that the evidence for valbenazine and deutetrabenazine suggests small therapeutic steps rather than large leaps forward and that, despite a known pharmacology, mode of benefit remains open to speculation. While any agent producing improvements in tardive dyskinesia is to be welcomed, the qualifications that must be imposed on bottom-line data are important, especially when one turns to perhaps the major issue in trial design and interpretation – power (Box 3). Although both trial programmes comprised impressively large samples in comparison to previous literature, it is questionable whether even they were adequately powered to address the primary question of efficacy, especially in expectation of slight change. One comprehensive review suggests that very large samples – in excess of 800 participants – would ideally be needed to evaluate properly drug interventions for tardive dyskinesia (Bergman Reference Bergman, Walker and Nikolakopoulou2017), implying sample sizes two to three times those for the pivotal valbenazine and deutetrabenazine studies. This may be impracticable and perhaps, with the key issue of masking systematically acknowledged, unnecessary but the point is crucial to interpretation and continues to support caution.
Non-pharmacological treatments
It is beyond the remit of this article and the knowledge requirements for psychiatrists to present details of physical treatments that have found advocacy in the management of hyperkinetic movement disorders and, by analogy, of tardive dyskinesia (Box 4). It is sufficient for psychiatrists to be aware that such interventions may be considered in consultation with neurology colleagues in intractable, incapacitating or life-threatening cases (Pouclet-Courtemanche Reference Pouclet-Courtemanche, Rouand and Thobois2016; Jankovic Reference Jankovic2017). Interestingly, botulinum toxin, occasionally useful, acts by cleaving SNAP-25, one of the SNARE family noted above, thereby preventing vesicular docking/release in peripheral cholinergic systems.
BOX 4 Physical treatments for tardive dyskinesia
Botulinum toxin
• Long-standing (>20 years) reports of benefit
• No systematic evaluation, including long-term outcomes
• Best for severe (focal) tardive dystonia
• Benefit may spread beyond target injection site
Deep-brain stimulation
• Invasive (but less so than lesioning)
• Globus pallidus interna is the focus in classic tardive dyskinesia
• Subthalamic nucleus is the target in tardive dystonia
• Potentially major issues with capacity/consent
• In extremis treatment only
Electroconvulsive therapy
• Scattered case/retrospective series reports only
• Benefit reported with ocular dystonia affecting vision
• Safe and familiar but use in tardive dyskinesia entirely empirical
• Benefits may be non-specific
• Probably best used only in those who also have conventional indications for ECT
Conclusions
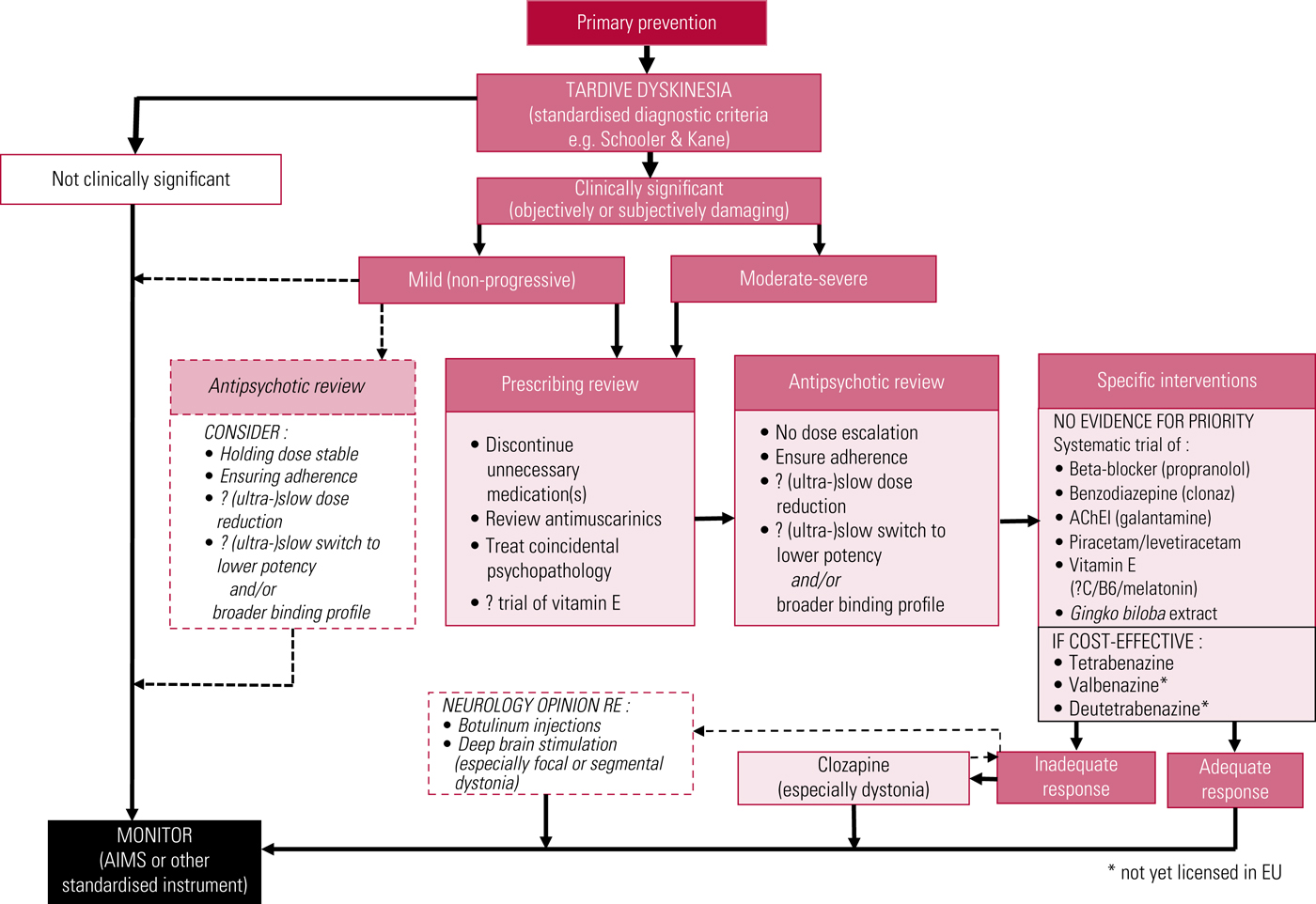
A proposed treatment algorithm is illustrated in Fig. 3, and American Academy of Neurology recommendations have recently been updated (Bhidayasiri Reference Bhidayasiri, Jitkritsadakul and Friedman2018).
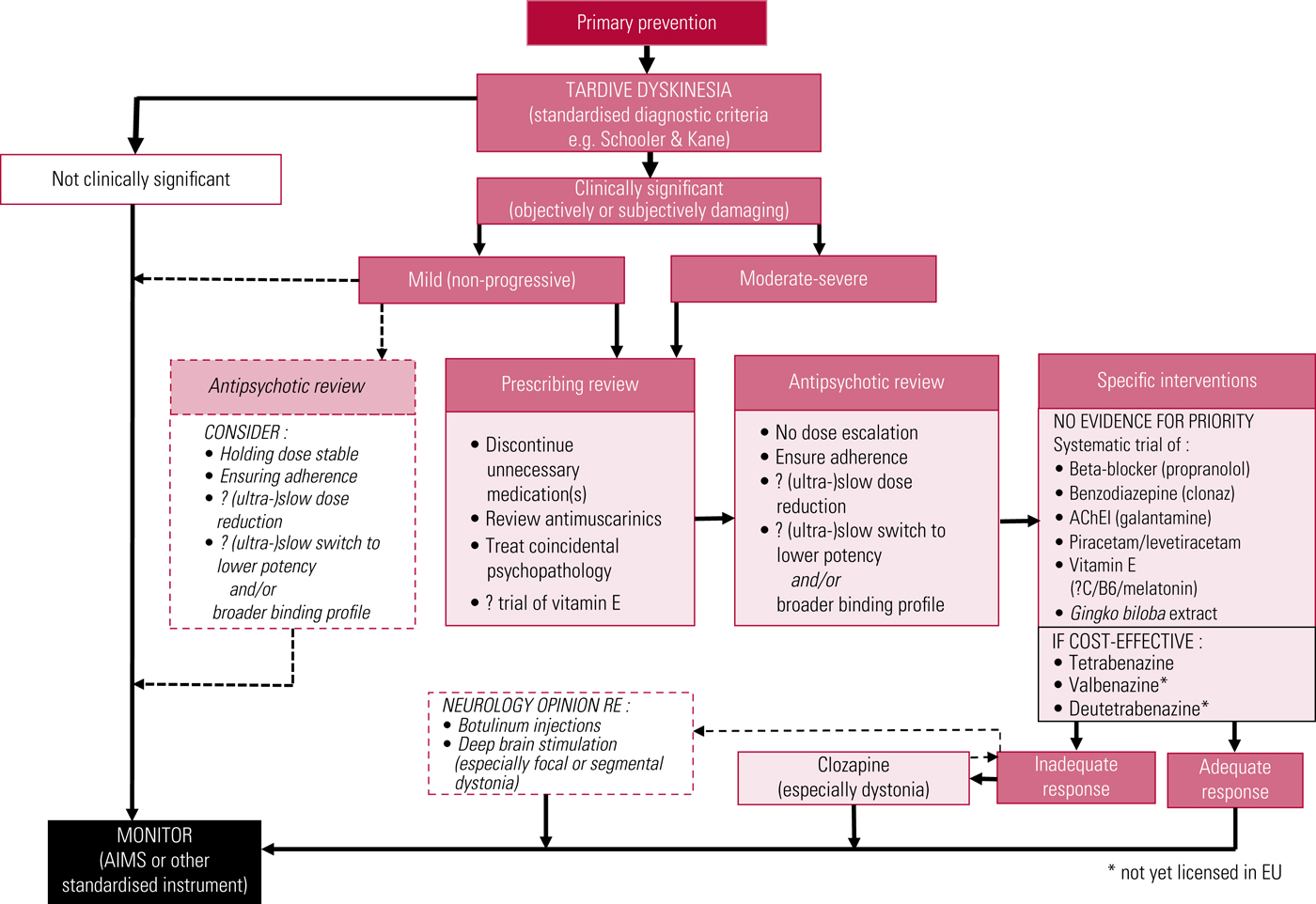
FIG 3 Proposed outline algorithm for treatment of tardive dyskinesia. AChEI, acetylcholinesterase inhibitor; AIMS, Abnormal Involuntary Movement Scale. After Owens (Reference Owens2014).
The treatment of tardive dyskinesia remains a challenge, no less than understanding its possibly varied causes. New treatments illustrate a welcome return of interest to a neglected area but in considering therapeutic options, prescribers must balance all the variables, including cost-effectiveness. Licensed drugs for the treatment of tardive dyskinesia remain expensive options for often modest gains.
MCQs
Select the single best option for each question stem
1 With regard to the aetiology of tardive dyskinesia:
a elevated CSF lipid peroxidase supports a role for free-radical excess in causation
b excess striatal acetylcholine transmission is the likely mediator of symptoms
c G-coupled protein changes following postsynaptic dopamine receptor blockade mediate oxidative stress
d glutamatergic involvement implicates ionotropic but not metabotropic receptors
e the primary imbalance emanates from excess dopaminergic activity in sensorimotor cortex.
2 Primary prevention of tardive dyskinesia includes:
a aiming for the maximum tolerated antipsychotic dose as quickly as possible
b antipsychotic dose reduction
c avoiding the use of antipsychotics as hypnotics
d coincidental treatment with an antimuscarinic drug as routine
e using only ‘atypical’ antipsychotics.
3 In relation to specific drug treatments recommended for tardive dyskinesia:
a acetylcholinesterase inhibitors are of no benefit
b benzodiazepines maintain their efficacy over the long term
c on the basis of the evidence base, valproate should be used more often
d the evidence supports early use of clozapine
e vitamin E (alpha-tocopherol) is an effective free-radical scavenger.
4 In relation to central neurotransmitter physiology:
a entrance of presynaptic cytosolic transmitter into vesicles is by simple diffusion
b each transmitter has its own transporter
c exocytosis is triggered by influx of chloride ions
d vesicular filling is facilitated by a proton pump
e VMAT2 comprises seven transmembrane domains.
5 Presynaptic depleting drugs:
a act by inhibiting the docking of vesicles onto the presynaptic membrane
b carry a significant suicide risk
c have been shown to impart particular benefits in oromandibular stereotypies
d have equal liability to cause drug-induced parkinsonism
e produce a conformational change in VMAT2.
MCQ answers
1 a 2 c 3 e 4 d 5 e






eLetters
No eLetters have been published for this article.