



Introduction
Fungi are probably the most widespread and diverse group of eukaryotic organisms inhabiting Antarctica, with a known fossil record dating back to the Permian period (White Jr & Taylor Reference White and Taylor1991, Harper et al. Reference Harper, Taylor, Krings and Taylor2016). They are involved in key processes in terrestrial ecosystems, such as decomposition and symbiotic mutualism (Treseder & Lennon Reference Treseder and Lennon2015, Asplund & Wardle Reference Asplund and Wardle2017), and therefore they contribute greatly to biogeochemical cycles in otherwise low-nutrient habitats. The number of known species in territories south of 60°S and archipelagos at lower latitudes, such as South Georgia, is ~1500 (Øvstedal & Lewis Smith Reference Øvstedal and Lewis Smith2001, Reference Øvstedal and Lewis Smith2011, Onofri et al. Reference Onofri, Selbmann, Zucconi, Tosi and de Hoog2005, Bridge et al. Reference Bridge, Spooner and Roberts2008, Bridge & Spooner Reference Bridge and Spooner2012). Almost a third of them associate symbiotically with eukaryotic algae (chlorophytes) or cyanobacteria, forming macroscopic lichen thalli. Lichens are in fact one of the most conspicuous elements of Antarctic terrestrial habitats, and their communities develop profusely in maritime areas (Søchting et al. Reference Søchting, Øvstedal and Sancho2004, Peat et al. Reference Peat, Clarke and Convey2007), and even in rocky outcrops at harsher locations in the continent (Kappen et al. Reference Kappen, Friedmann and Garty1981, Broady & Weinstein Reference Broady and Weinstein1998, Pérez-Ortega et al. Reference Pérez-Ortega, Ortiz-Álvarez, Green and de los Ríos2012). On the other hand, non-lichenized fungi generally remain unnoticeable because either they are unicellular (e.g. yeasts) or they form unseen mycelia and small reproductive structures. Microfungi are the most abundant in Antarctic soils (Vishniac & Hempfling Reference Vishniac and Hempfling1979, Ruisi et al. Reference Ruisi, Barreca, Selbmann, Zucconi and Onofri2007, Arenz & Blanchette Reference Arenz and Blanchette2011, Arenz et al. Reference Arenz, Blanchette, Farrell and Cowan2014), and many Ascomycota and even some Basidiomycota have been reported on wood brought to Antarctica (Arenz & Blanchette Reference Arenz and Blanchette2009, Blanchette et al. Reference Blanchette, Held, Arenz, Jurgens, Baltes, Duncan and Farrell2010, Arenz et al. Reference Arenz, Held, Jurgens and Blanchette2011, Held & Blanchette Reference Held and Blanchette2017). Additional research using next-generation sequencing techniques has also revealed a high diversity of fungi even in the most unexpected habitats (Coleine et al. Reference Coleine, Stajich, Zucconi, Onofri, Pombubpa and Egidi2018, Garrido-Benavent et al. Reference Garrido-Benavent, Pérez-Ortega, Durán, Ascaso, Pointing and Rodríguez-Cielos2020, Rosa et al. Reference Rosa, Pinto, Convey, Carvalho-Silva, Rosa and Câmara2021), with similarities in species diversity and community composition to the Arctic (Cox et al. Reference Cox, Newsham, Bol, Dungait and Robinson2016).
Macrofungi (i.e. non-lichenized species that form relatively large fruiting bodies or ‘mushrooms’) are infrequently reported in Antarctica, with a few dozen species growing in the climatically milder sub-Antarctic, Maritime Antarctica and occasionally in the western Antarctic Peninsula, often occurring on large carpets of mosses and vascular plants (Pegler et al. Reference Pegler, Spooner and Lewis Smith1980, Gumińska et al. Reference Gumińska, Heinrich and Olech1994, Bridge et al. Reference Bridge, Spooner and Roberts2008, Putzke et al. Reference Putzke, Putzke, Pereira and Albuquerque2012, Held & Blanchette Reference Held and Blanchette2017, Newsham et al. Reference Newsham, Davey, Hopkins and Dennis2021). Bridge & Spooner (Reference Bridge and Spooner2012) and Newsham et al. (Reference Newsham, Davey, Hopkins and Dennis2021) suggested that the general absence of large land animals and higher plants with woody components in Antarctica is a limiting factor for the development of these fungi. From a taxonomic and biogeographical viewpoint, the scarcity of available collections of mushroom-forming fungi from the Antarctic has so far impeded detailed comparisons with species that are known from elsewhere. More specifically, the lack of genetic data has prevented the deciphering of the most probable temporal and spatial origins of the Antarctic populations of certain species. In fact, despite being one of the most diverse groups in the whole continent, there is still a general lack of knowledge as to whether non-lichenized fungi, and particularly the Antarctic macrofungi, form specific populations of cosmopolitan species or constitute true endemic species (Bridge & Spooner Reference Bridge and Spooner2012, Arenz et al. Reference Arenz, Blanchette, Farrell and Cowan2014).
To provide an answer to this question, the present work uses recent collections of fruiting bodies of Galerina species to assess their taxonomic identity and estimate a date for their Antarctic origin within a phylogenetic framework. This genus of basidiomycetous fungi encompasses ~300 species worldwide (Horak Reference Horak1994, Gulden et al. Reference Gulden, Stensrud, Shalchian-Tabrizi and Kauserud2005), which form relatively small, yellowish to reddish-brown fruiting bodies with campanulate, convex to flat pilei and slender stipes. Several Galerina species are well known for posing a poisoning risk due to the production of deadly amatoxins (Landry et al. Reference Landry, Whitton, Bazzicalupo, Ceska and Berbee2021). The genus shows a broad distribution in Mediterranean, temperate and boreal regions in the Northern Hemisphere (GBIF 2022), where saprotrophic species generally grow on dead parts of bryophytes in peat bogs or are associated with woody remnants or other plant debris in forests, on which this genus degrades wood cell wall components (Gulden et al. Reference Gulden, Stensrud, Shalchian-Tabrizi and Kauserud2005, Grzesiak & Wolski Reference Grzesiak and Wolski2015, Kohler et al. Reference Kohler, Kuo, Nagy, Morin, Barry and Buscot2015). In the Antarctic continent and nearby archipelagos, the number of Galerina species reported is ~11, with Galerina antarctica Singer, Galerina glebarum (Berk.) Singer and Galerina perrara Singer originally being described, and these are known only from these regions (Fig. 1; Berkeley Reference Berkeley and Hooker1847, Singer & Corte Reference Singer and Corte1962, Pegler et al. Reference Pegler, Spooner and Lewis Smith1980, Bridge et al. Reference Bridge, Spooner and Roberts2008). Based on the inferred phylogeny, we aim to ascertain whether the sequenced Antarctic specimens belong to geographically restricted, species-level lineages (i.e. putative endemic species) or conform to particular intraspecific lineages of cosmopolitan Galerina (non-endemic species). In lichenized fungi, Antarctic endemic species have been shown to have a relictual, pre-Pleistocene origin, whereas Antarctic populations of amphitropical lichens are generally much younger, dating back from the Pleistocene onwards (Fernández-Mendoza & Printzen Reference Fernández-Mendoza and Printzen2013, Garrido-Benavent et al. Reference Garrido-Benavent, Søchting, De Los Ríos Murillo and Pérez-Ortega2016, Reference Garrido-Benavent, De Los Ríos, Fernández-Mendoza and Pérez-Ortega2018, Reference Garrido-Benavent, Pérez-Ortega, De Los Ríos, Mayrhofer and Fernández-Mendoza2021). The temporal frame estimated with the time-calibrated Galerina phylogeny will further help us to discern whether their evolution in Antarctica conforms to either of these two scenarios.

Fig. 1. Diversity and distribution of Galerina species in Antarctica based on collection data provided by Bridge et al. (Reference Bridge, Spooner and Roberts2008), Arenz et al. (Reference Arenz, Blanchette, Farrell and Cowan2014), Krishnan et al. (Reference Krishnan, Convey, González-Rocha and Alias2016) and Canini et al. (Reference Canini, Geml, D'Acqui, Selbmann, Onofri, Ventura and Zucconi2020). Species for which samples have been included in the present work are in bold.
Material and methods
Fieldwork and morpho-anatomical study of fruiting bodies
Several fruiting bodies of Galerina growing in a localized area, and therefore probably corresponding to a single mycelium, were collected in March 2018 from Livingston Island (South Shetland Islands) and more specifically in Punta Hannah (62°39‘15.37″ S, 60°36’27.44″ W, 377 m above sea level), which is the second largest island in the South Shetland Islands, a mountainous archipelago located in Maritime Antarctica. These fruiting bodies grew abundantly on soil, with a profuse development of cryptogams (mosses and the chlorophyte macroalgae Prasiola) and the Antarctic hair grass Deschampsia antarctica Desv. (Fig. 2). Sampling permit no. CPE-2017-3 was obtained through the Spanish Polar Committee. Specimens were frozen until further processing at the laboratory, where they were observed under a Leica S8APO dissecting microscope equipped with a Leica EC3 image capture system. Handmade sections of lamellae were rehydrated in distilled H2O to describe anatomical characteristics. Microscopic observations were made using a Zeiss Axioplan 2 microscope fitted with ‘Nomarski’ differential interference contrast, and photographs were taken with a Zeiss AxioCam digital camera. Microscopic measurements were made by means of the Zeiss Axiovision 4.8 imaging system. Reported data are averages followed by standard deviations, and the maximum and minimum values are given in parentheses.

Fig. 2. Galerina marginata: a. fruiting bodies growing on a carpet of Deschampsia antarctica and Prasiola sp. in Punta Hannah (Livingston Island), b. a detail of the fruiting body pileus, c. a basidium (i.e. basidiomycete sporangium) with developing spores and d. spores. Scale bars: 10 μm.
DNA extraction and polymerase chain reaction amplification
The isolation of genomic DNA from a single Galerina basidioma (pl. basidiomata; i.e. basidiomycete fruiting bodies) was done from a piece of lamellae and using the Speed Tools DNA Extraction Kit (Biotools, Madrid, Spain), following the manufacturer's recommendations. The extracted DNA was eluted in a final volume of 60 μl with sterile purified water (SIGMA). Sequence data of the internal transcribed spacer of the nuclear ribosomal DNA (the so-called fungal barcode marker; Schoch et al. Reference Schoch, Seifert, Huhndorf, Robert, Spouge and Levesque2012) was amplified using the primer pair ITS1F-KYO2 and ITS4-KYO2 (Toju et al. Reference Toju, Tanabe, Yamamoto and Sato2012). Polymerase chain reaction (PCR) experiments were performed in a total volume of 10 μl, containing 1 μl of reaction buffer (Biotools®), 2 μl of dNTPs (1 mM), 0.5 μl of each primer (10 μM), 0.2 U of DNA polymerase (Biotools®) and 1.5 μl of the genomic DNA elution; the final volume was reached by adding distilled water (SIGMA). The following PCR temperature profile was employed: 5 min at 95°C, then 30 cycles of 30 s at 95°C, 1 min at 52°C and 1.5 min at 72°C, with a final extension of 10 min at 72°C. The PCR experiments were visualized on 1% agarose gel stained with PRONASAFE nucleic acid stain solution (CONDA Laboratories). The PCR products were purified and cleaned using the UltraClean PCR Clean-Up Kit (MOBIO Laboratories, Inc.). Both complementary DNA strands were sequenced at Macrogen Europe (Spain) using the same primer set as for the initial amplification. Electropherograms were checked and assembled using SeqManII v.5.07© (DNASTAR, Inc.).
Compilation of the specimen-based nrITS dataset and sequence alignment
The newly produced sequence was submitted to the BLAST online tool (Altschul et al. Reference Altschul, Gish, Miller, Myers and Lipman1990) to check for possible PCR product contamination and to identify and retrieve available, highly similar nrITS sequences. To this purpose, the GenBank (http://www.ncbi.nlm.nih.gov/), UNITE (Nilsson et al. Reference Nilsson, Larsson, Taylor, Bengtsson-Palme, Jeppesen and Schigel2019) and BOLD (Ratnasingham & Hebert Reference Ratnasingham and Hebert2007) nucleotide databases were used as references. A total of 118 sequences (97 GenBank, 13 UNITE and 8 BOLD) spanning a 97–100% similarity range were downloaded. Most were accessions labelled with the species name Galerina marginata (Batsch) Kühner. A closely related nrITS sequence of a Galerina collection from Amsler Island (Antarctic Peninsula) was included as well. This was also collected from an area where mosses were growing under permit ACA-2012-013. DNA extraction and sequencing were done using methods previously described (Blanchette et al. Reference Blanchette, Held, Hellmann, Millman and Büntgen2016). An additional search in public databases was conducted to select and retrieve any available nrITS data for other Antarctic Galerina collections. Two sequences obtained from soil isolates were found: MK537266, which was generated in a study by Canini et al. (Reference Canini, Geml, D'Acqui, Selbmann, Onofri, Ventura and Zucconi2020) from Victoria Land; and MF692967, from King George Island (Krishnan et al. Reference Krishnan, Convey, González, Smykla and Alias2018). BLAST searches against the GenBank database revealed a close match of these accessions to nrITS sequences labelled with the species names Galerina badipes (Pers.) Kühner and Galerina fallax A.H. Sm. & Singer. Twenty-five sequences hosted in GenBank belonging to these two species were downloaded and incorporated into the dataset as well. Finally, the dataset was completed by including additional Galerina species based on works by Gulden et al. (Reference Gulden, Stensrud, Shalchian-Tabrizi and Kauserud2005) and Latha et al. (Reference Latha, Raj, Paramban and Manimohan2015), which provided two of the most comprehensive Galerina phylogenies published to date. The final nrITS consisted of 178 sequences. We followed Gulden et al. (Reference Gulden, Stensrud, Shalchian-Tabrizi and Kauserud2005) in selecting adequate outgroup taxa for our phylogenetic analyses. Although the genus Galerina was revealed to be polyphyletic by these authors, they considered a group of taxa referred to ‘tubariopsis’ to be a suitable outgroup. In our dataset, this is represented by the following species: Galerina arctica (Singer) Nezdojm., Galerina clavata (Velen.) Kühner, Galerina discreta E. Horak, Senn-Irlet, M. Curti & Musumeci, Galerina laevis Singer, Galerina pseudocerina A.H. Sm. & Singer and Galerina stordalii A.H. Sm.
The program MAFFT v.7.308 (Katoh & Standley Reference Katoh and Standley2013) was used to generate a multiple-sequence alignment with the following parameters: the FFT-NS-I x1000 algorithm, the 200PAM/k = 2 scoring matrix, a gap open penalty of 1.5 and an offset value of 0.123. The resulting alignment was manually optimized in Geneious v.9.0.2 to 1) trim alignment ends of longer sequences that included part of the 18S–28S ribosomal subunits, 2) replace gaps at the ends of shorter sequences with an International Union of Pure and Applied Chemistry (IUPAC) base representing any base (‘N’) and 3) replace doubtful base calls at the extremes with ‘N’. The software GBlocks 0.91b (Castresana Reference Castresana2000) was subsequently used to automatically deal with ambiguously aligned regions, implementing the least stringent parameters but allowing gaps in 50% of the sequences. Alignments were deposited in FigShare (DOI: 10.6084/m9.figshare.22219546).
Maximum-likelihood phylogenetic analyses
The online version of RAxML-HPC2 hosted at the CIPRES Science Gateway (Stamatakis Reference Stamatakis2006, Stamatakis et al. Reference Stamatakis, Hoover and Rougemont2008, Miller et al. Reference Miller, Pfeiffer and Schwartz2010) was used to estimate two maximum-likelihood (ML) phylogenies based on the GBlocks-trimmed (GB) and untrimmed (ORG) alignments. This approach would allow us to evaluate the effect of alignment uncertainty on the inferred nodal support. The analyses used the GTRGAMMA nucleotide substitution model for the two delimited partitions within the nrITS (ITS1+2, 5.8S), and nodal support was evaluated by conducting 1000 rapid bootstrap pseudoreplicates. The resulting phylogenetic trees were visualized in FigTree v.1.4 (http://tree.bio.ed.ac.uk/software/tracer/), and Adobe Illustrator CS5 was used for artwork. Tree nodes with bootstrap support (BS) values ≥ 70% were regarded as significantly supported.
Haplotype networks, DNA polymorphism and neutrality tests
The genealogical relationships among specimens included in the G. marginata clade were calculated under a statistical parsimony framework in PopART v.1.7 (Leigh & Bryant Reference Leigh and Bryant2015) using the method of Templeton et al. (Reference Templeton, Crandall and Sing1992). To this purpose, a sub-alignment of 120 sequences was extracted from the GBlocks-untrimmed, original alignment. Because the inference of haplotype networks is sensitive to ambiguous base calls and missing data (Joly et al. Reference Joly, Stevens and van Vuuren2007), the sub-alignment was edited to remove 16 sequences with a high proportion of missing data at their extremes and 34 sequences with ambiguous base calls occurring at polymorphic positions. Haplotypes were subsequently inferred with DnaSP v.5.10 (Librado & Rozas Reference Librado and Rozas2009) considering sites with alignment gaps and removing invariable sites. The network was artistically edited in Adobe Illustrator CS5 and haplotypes were labelled according to their geographical origin. DNA polymorphism in the 70 remaining sequences was evaluated with the software DnaSP v.5.10 (Librado & Rozas Reference Librado and Rozas2009). The computed indices were the number of segregating sites (s), the number of haplotypes (h), haplotype diversity (Hd) calculated without considering gap positions and the nucleotide diversity (π) using the Jukes & Cantor (Reference Jukes and Cantor1969) correction. Deviations from neutrality with Tajima's D and Fu's Fs statistics were also assessed to infer past population size changes. The tests were carried out in DnaSP v.5.10 using the number of segregating sites, and their significance was assessed based on 104 coalescent simulations. We did not infer haplotype networks, nor do we evaluate DNA polymorphism for G. badipes and G. fallax because of the few sequences these species encompassed and due to the substantial amount of missing data in sequences masking the existing polymorphism.
Dating analyses
The inference of a time frame for the global evolutionary history of Galerina was conducted under a Bayesian framework with BEAST 1.8.1 (Drummond et al. Reference Drummond, Suchard, Xie and Rambaut2012). Because this Basidiomycota genus lacks a suitable fossil record, the dating analysis used a secondary calibration imposed on the nrITS substitution rate. Hence, the BEAST analysis implemented the average rate of 4.61 × 10-3 substitutions per site per million years (s/s/Ma) inferred for the genus Phaeocollybia R. Heim in Ryberg & Matheny (Reference Ryberg and Matheny2012), because this genus and Galerina belong into the family Hymenogastraceae (Matheny et al. Reference Matheny, Moreau, Vizzini, Harrower, De Haan, Contu and Curti2015). This analysis was referred to as Dating A. To take into account the uncertainty associated with that rate, we re-ran analyses using the estimates representing the minimum (2.92 × 10-3 s/s/Ma, Dating B) and maximum (6.45 × 10-3 s/s/Ma, Dating C) values of the rate's 95% credibility interval provided by Ryberg & Matheny (Reference Ryberg and Matheny2012). The analyses were conducted with the two alignment versions (GB and ORG) to learn about the impact on age estimates of keeping ambiguously aligned positions in the alignment. Redundant sequences were removed from the alignments using the FaBox v.1.41 online toolbox (Villesen Reference Villesen2007). PartitionFinder 1.1.1 (Lanfear et al. Reference Lanfear, Calcott, Ho and Guindon2012) was used to infer optimal substitution models for the two nrITS partitions considering a model with linked branch lengths and the Bayesian information criterion. This analysis favoured the GTR+Γ model for the ITS1+ITS2 partition and the K80 model for the 5.8S partition. We conducted preliminary Bayes factor comparisons (Kass & Raftery Reference Kass and Raftery1995) of ML estimates (MLEs) calculated with path sampling and stepping-stone approaches (Lartillot & Philippe Reference Lartillot and Philippe2006, Xie et al. Reference Xie, Lewis, Fan, Kuo and Chen2011) to choose among different BEAST tree priors and molecular clocks. The use of an uncorrelated lognormal relaxed molecular clock over the strict clock was strongly supported for the GB and ORG datasets (Tables SI & SII). As for the tree priors, models incorporating the coalescent-constant size produced substantially higher MLE values than models using the birth-death and Yule process priors. Runs using chain lengths of 1.5 × 108 steps were implemented, and parameters were logged every 1.5 × 104 steps. Resulting log files were checked in Tracer 1.7 to ensure that all parameters had effective sample sizes > 200 after removing the first 20% of saved trees as burn-in. Then, the median heights of the 1 × 104 post-burn-in tree samples were annotated with TreeAnnotator 1.8.1, and the chronograms were drawn with FigTree 1.4. Tracer 1.7 and TreeAnnotator 1.8.1 are available at http://tree.bio.ed.ac.uk/. We set the value of Bayesian posterior probabilities (PPs) at a minimum of 0.97 for considering tree nodes to be well supported.
Results
Specimen study
The G. marginata specimens studied morphologically and phylogenetically showed relatively small pilei of up to 4 cm in diameter, first convex and then turning flat (Fig. 2). Basidia were tetrasporic and produced ellipsoid to broadly amygdaloid, brownish and verrucose spores, with a rounded apex and a visible hilar appendix and containing one to two guttules. The size (length × width) of 25 spores measured in water was (11.2) 13.0 ± 1.0 (15.8) × (6.2) 7.4 ± 0.8 (9.9) μm, and the length/width ratio was (1.4) 1.8 ± 0.1 (2.0). Care must be taken when comparing the size of pilei and microscopic characteristics with literature data because specimens were brought back from Antarctica frozen and were examined after melting, a process that might have affected the structure of these characteristics.
Alignments and phylogenies under ML
The original alignment (ORG) done with MAFFT consisted of 178 Galerina nrITS sequences and 665 positions, of which 279 were variable and 82 corresponded to singleton sites. After processing the alignment with GBlocks (GB), 603 positions (90% of the original alignment) were retained in 29 selected blocks; 255 positions were variable and 73 were singleton sites. The ML analyses in RAxML estimated phylogenies with lnL = -5168.2 (GB) and lnL = -5493.96 (ORG). Although the topologies inferred based on the two alignments were not identical, they showed no supported conflicts (Supplemental Figs 1 & 2). In general, sister relationships among the different Galerina species included in the ingroup lacked support. Bootstrap values > 70% were obtained for the crown nodes encompassing all G. marginata, G. badipes and G. fallax sequences, where the data obtained from Antarctic material are placed in both topologies. It must be highlighted that the G. marginata clade, hereinafter referred to as G. marginata s.l., included sequences from specimens originally labelled as Galerina autumnalis, G. hygrophila, G. pseudomycenopsis, G. unicolor and G. venenata (including its type sequence, MH827070). Furthermore, BS values > 78% were found for the sister relationship of G. marginata s.l. and G. badipes, G. minima and G. atkinsoniana, G. pseudobadipes and G. stylifera, and G. cephalotricha and G. mniophila. The sister relationship of G. jaapii with the clade containing G. marginata s.l. and G. badipes received a BS value of 82% (GB), whereas the clade containing the latter three species along with G. indica had a BS value of 81%. Within G. marginata s.l., the newly generated sequence from Livingston Island (GenBank accession OQ569484) was located at the bottom and close to three other sequences from Antarctica: OP795715, which was obtained from a basidioma collected at Norsel Point on Amsler Island and differed by one nucleotide; KU559684, an environmental sequence co-occurring in Antarctica and the Arctic (Cox et al. Reference Cox, Newsham, Bol, Dungait and Robinson2016), which is shorter than the other sequences and therefore was composed of a number of missing nucleotides; and KT990212, labelled as ‘Arrhenia antarctica’ and collected by Halina Galera from an uncertain location within Antarctica that showed missing and ambiguous positions together with one diverging nucleotide. Two sequences labelled as G. pseudomycenopsis (AJ585503 and GU234057) collected in the USA and Svalbard were closely related as well and differed, in general, by fewer than five alignment positions.
Genetic diversity in G. marginata s.l.
Forty-two haplotypes, producing a haplotype diversity (Hd) of 0.930, were recovered from the 70 analysed sequences of G. marginata s.l. (Fig. 3). The haplotype network revealed a close relationship between the two Antarctic haplotypes and others obtained from Northern Hemisphere specimens, including either North America (USA and Canada) or northern Europe (Scandinavia, Baltic countries and the UK). Single mutations segregated these haplotypes. Furthermore, the haplotype network showed two star-like sub-networks separated by just one mutation. The most evident sub-network was composed of a central haplotype with a wide distribution in North America (especially in Canada) that also occurred in Switzerland (central Europe). Connected to this central haplotype by just one or two mutations were several minor haplotypes from North America and northern Europe. In contrast, the second star-like sub-network had a central haplotype distributed in Europe overall and the Caucasus mountainous region, and this was linked to minor haplotypes distributed in Europe as well. At the bottom of the network in Fig. 3, a number of haplotypes from Asia (Altay Republic, China, South Korea and Japan) were connected by one or up to four mutations to haplotypes occurring in North America and Mexico. A couple of these haplotypes were shared by regions on both sides of the Pacific Ocean. Finally, Tajima's and Fu's neutrality tests estimated negative values of D (-1.46358, P > 0.10; not significant) and Fs (-24.755, P < 0.001; significant). The negative values of D and Fs indicate an excess of low-frequency polymorphisms relative to expectation and an excess of the number of alleles, respectively. Collectively, the results from these two tests suggest a population or demographic expansion in G. marginata s.l.

Fig. 3. Statistical parsimony networks connecting Galerina marginata haplotypes and summary of DNA polymorphism indices. Haplotypes were coloured according to the geographical origin of samples. The sizes of the circles in the networks are proportional to the numbers of individuals bearing the haplotype; black-filled smaller circles indicate missing haplotypes. Mutations are shown as hatch marks. S = segregating sites; h = number of haplotypes; Hd = haplotype diversity; π = nucleotide diversity.
BEAST phylogenies and age estimates
The average effective sample sizes were > 200 for all parameters in the Bayesian dating analyses conducted with BEAST based on the two alignments (GB and ORG). No supported topological conflicts were observed between the two BEAST topologies (Fig. 4) or between them and those obtained in RAxML under ML (Supplemental Figs 1 & 2). However, the BEAST analysis using the GB alignment found support (PP > 0.97) for a sister relationship between a Galerina sp. collected from the Balearic Islands (MH817980) and the bulk of G. marginata s.l. sequences. Within G. marginata s.l., sequences from Antarctica were phylogenetically close to the same sequences reported in the RAxML analyses. Moreover, two highly supported clades were revealed: one encompassing several sequences from North America and Asia, which correspond with the haplotypes shown at the bottom of the haplotype network (Fig. 3); and a second clade containing several sequences obtained from North American as well as one from Switzerland. This clade is represented by the most evident star-like sub-network in Fig. 3. High support was also revealed for a clade containing G. marginata s.l. and G. badipes together with G. jaapii, G. indica, G. triscopa f. telamonioides, G. pseudocamerina, G. pruinatipes, G. chionophila and G. nana. Supported sister relationships were also found for G. calyptrata, G. sphagnicola and G. luteolosperma and for the pair G. mniophila-cephalotricha.
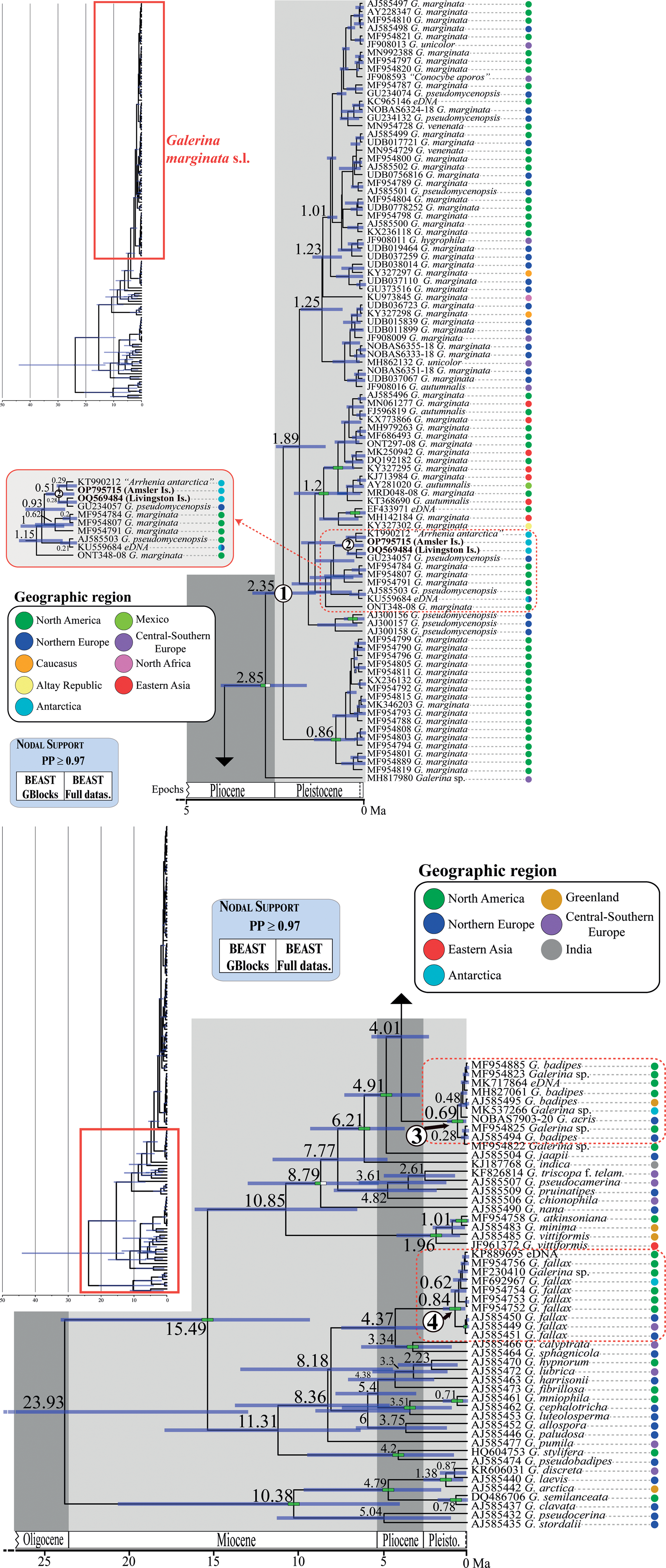
Fig. 4. Chronogram obtained with BEAST based on nrITS data depicting the evolutionary history of Galerina species. Dashed red rectangles highlight the clades where the Antarctic collections are included. The mean age estimate for the divergence of selected nodes is provided in million years ago (Ma). For each terminal in the tree, the GenBank, UNITE or BOLD nrITS accession number, the taxonomic identity as originally deposited in these databases and the geographical origin are given. Green-filled rectangles indicate nodal support (posterior probability (PP) ≥ 0.97) in analyses using the two versions of the nrITS alignment (GB and ORG). The newly produced nrITS sequences with the corresponding GenBank codes are highlighted in bold. Numbers 1–4 in white circles indicate phylogenetic clades where sequenced specimens of Antarctic Galerina are placed: 1–2 = Galerina marginata, 3 = Galerina badipes) and 4 = Galerina fallax.
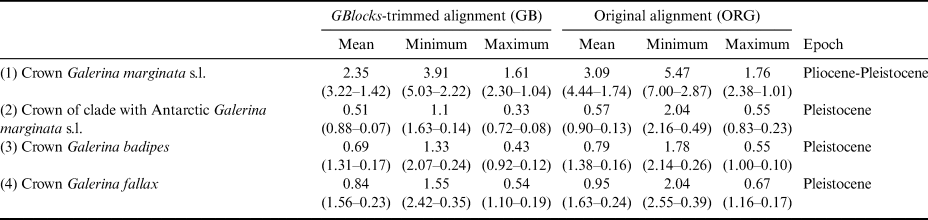
The dating analysis based on the GB alignment generated slightly lower age estimates than the analysis that used the ORG alignment (Table I). However, the inferred 95% highest posterior density (HPD) intervals obtained with the two alternative analyses overlapped to a considerable extent (Table I). For example, when the average nrITS substitution rate of 4.61 × 10-3 s/s/Ma was employed (Dating A), the crown node of G. marginata s.l. was dated back to 2.35 Ma (3.22–1.42 Ma, 95% HPD; GB alignment) and 3.09 Ma (4.44–1.74 Ma, 95% HPD; ORG alignment), a time interval at the transition from the Pliocene to the Pleistocene. For simplicity, the discussion below is based on the chronogram estimated with the GB alignment (Fig. 4) because it did not include potentially misaligned (ambiguous) regions. This chronogram reveals that major diversification events in Galerina took place since the Miocene epoch (ca. 23.03 to 5.30 Ma) and extended into the Pliocene (ca. 5.30 to 2.58 Ma). Speciation occurred also in the Pleistocene (ca. 2.58 Ma to 11 700 years ago), as is observed between the species G. mniophila and G. cephalotricha and between G. minima and G. atkinsoniana. Intraspecific diversification in G. marginata s.l., G. badipes and G. fallax, which include sequences obtained from Antarctic material, occurred mainly in the Pleistocene, with the Antarctic haplotypes originating during the last 500 000 years on average (Fig. 4). Finally, it should be highlighted that Dating B and C, which used the minimum and maximum values of the rate's 95% credibility interval provided by Ryberg & Matheny (Reference Ryberg and Matheny2012), produced older and younger age estimates, respectively, compared with results using the average rate value. Table I summarizes the age estimates and corresponding 95% HPD intervals for selected nodes (see Fig. 4) based on the three dating analyses (Dating A, B and C).
Table I. Estimated divergence ages for the selected crown nodes in Fig. 4 representing Galerina species with Antarctic populations. The dating analyses in BEAST used alternative nrITS alignment versions (GB vs ORG). For each of these versions, the mean age value and the corresponding 95% highest posterior density intervals are provided in million years ago (Ma) considering the mean, minimum or maximum values of the nrITS substitution rate inferred for the genus Phaeocollybia in Ryberg & Matheny (Reference Ryberg and Matheny2012), which was used here for calibration purposes. The time frame proposed in the ‘Epoch’ column considered the six mean ages estimated in each row.
Discussion
The present study validated by means of molecular phylogenetics the existence in Antarctica of populations of G. marginata, G. badipes and G. fallax. The former species (and probably G. badipes too) is well known for producing amatoxins, which can have dramatic consequences for human ingestion (Landry et al. Reference Landry, Whitton, Bazzicalupo, Ceska and Berbee2021). The samples of Galerina collected from Amsler Island, Antarctica, were also found to contain alpha-amanitin (unpublished data 2013, analyses completed by Jonathan Walton, University of Michigan). These three Galerina species, G. marginata, G. badipes and G. fallax, represent relatively common macrofungi in Mediterranean and Temperate-Arctic ecosystems in the Northern Hemisphere (GBIF 2022). The closest genetic lineages to sequenced Antarctic Galerina were in fact collected in northern Europe (G. marginata), Greenland (G. badipes) and North America (G. fallax), according to the inferred phylogenies. Therefore, their distribution is here shown to be potentially sub-cosmopolitan or amphitropical, given the few occurrences in tropical regions (GBIF 2022). This biogeographical interpretation of our results supports the opinion of Pegler et al. (Reference Pegler, Spooner and Lewis Smith1980), who used morphological evidence to suggest a close similarity between macrofungal species from the sub-Antarctic and the Temperate-Arctic regions of the Northern Hemisphere. The distribution patterns of the studied Galerina match to a great extent with the global geographical distribution of some non-lichenized Antarctic microfungi (Bridge & Newsham Reference Bridge and Newsham2009, Bridge & Spooner Reference Bridge and Spooner2012, Cox et al. Reference Cox, Newsham, Bol, Dungait and Robinson2016), but, most interestingly, they also match with the amphitropical distribution pattern displayed by a significant proportion of lichenized fungi, which in Antarctica account for almost 40% of lichens (Øvstedal & Lewis Smith Reference Øvstedal and Lewis Smith2001). The existence of nearly identical global distribution patterns in various lichenized and non-lichenized Antarctic fungi makes us hypothesize that, at the geological time scale, these species overcame similar ecological and geographical filters to acquire their current distribution. To accumulate evidence for supporting or rejecting this hypothesis, other Antarctic species of Galerina and members of additional non-lichenized macrofungi genera should be surveyed and studied phylogenetically.
The phylogenetic and haplotype network analyses indicated that the studied intraspecific genetic lineages of the Antarctic Galerina might be geographically restricted and therefore endemic to this polar region. The fact that the considered species produced fruiting bodies in the surveyed Antarctic localities indicates that these macrofungi have established permanent populations, and therefore they are not transient visitors. Basidiomata formation represents the last step in the life cycle of Basidiomycota fungi. Briefly, it starts with spore germination and mycelium growth once abiotic and biotic requirements are met, followed usually by mating of two compatible, distinct mycelia, and, as a result, basidiomata develop and spores are produced and released after meiosis. We suggest that the three Galerina species have been established in this region long enough for mutations to accumulate in the studied genetic locus, the nrITS, which is known to evolve at a higher rate compared to other commonly used fungal molecular markers (Schoch et al. Reference Schoch, Seifert, Huhndorf, Robert, Spouge and Levesque2012). Moreover, the existence of Antarctic-endemic intraspecific lineages of these fungi is of the utmost importance for designing conservation policies that consider a broad spectrum of eukaryotic organisms and not only plants and animals.
However, assessing endemicity in fungi poses some risks, even at the intraspecific genetic level. In fact, the existence or not of true Antarctic-endemic non-lichenized fungal lineages has been hotly debated (e.g. Bridge & Spooner Reference Bridge and Spooner2012, Arenz et al. Reference Arenz, Blanchette, Farrell and Cowan2014) because of the obvious difficulties in observing and/or isolating macro- and microfungi in Antarctica, or elsewhere, due to their complex life cycles, ecologies and/or sizes. In this sense, Bridge & Spooner (Reference Bridge and Spooner2012) mentioned that some of the allegedly Antarctic-endemic species reported by Onofri et al. (Reference Onofri, Selbmann, Zucconi, Tosi and de Hoog2005) were found later elsewhere. Assessing endemicity in lichenized fungi is comparatively more straightforward because they usually form macroscopic and enduring lichen thalli (but see Hale et al. Reference Hale, Fisher, Keuler, Smith and Leavitt2019). Hence, the proportion of Antarctic-endemic lichens has been estimated at ~30% (Øvstedal & Lewis Smith Reference Øvstedal and Lewis Smith2001). In our opinion, the analysis of biogeographical patterns in non-lichenized Antarctic fungi may be more accurate if approached phylogenetically as long as extensive specimen and molecular datasets are compiled. For example, genotypes restricted to Antarctica were revealed for widespread fungi, such as the ascomycete Thelebolus microsporus (Berk. & Broome) Kimbr. and other microfungi (de Hoog et al. Reference de Hoog, Gottlich, Platas, Genilloud, Leotta and van Brummelen2005, Bridge & Newsham Reference Bridge and Newsham2009, Bridge & Spooner Reference Bridge and Spooner2012, Gonçalves et al. Reference Gonçalves, Vitoreli, de Menezes, Mendes, Secchi, Rosa and Rosa2017). Even so, the lack of availability of sequence data and collections from as yet unexplored areas in the Southern Hemisphere makes it difficult to assess endemicity in Antarctic fungi. Although we compiled a large specimen dataset in the present Galerina study, it lacked sequence data associated with reports of G. marginata and G. badipes from Australia and New Zealand (GBIF 2022). Because of the geographical proximity of these austral regions to Antarctica, it would be worth checking whether Antarctic, Australian and New Zealand populations of Galerina share the same genotype or are at least closely related. In this way, endemicity or colonization routes would be judged more correctly. Furthermore, DNA sequence length and quality are crucial for precise interpretations of biogeographical patterns. This is of particular importance today due to the abundance of short sequences from DNA metabarcoding studies. If short and long sequences are combined in datasets, the resulting biogeographical interpretations could be misleading to some extent, as unrealistic phylogenetic affinities could be inferred. For example, we used a short nrITS sequence of G. marginata that co-occurred in Antarctica and the Arctic (KU559684; Cox et al. Reference Cox, Newsham, Bol, Dungait and Robinson2016). Despite the existence of identical DNA sequences in individuals from both poles having been observed in, for example, the lichenized fungus Pseudephebe minuscula (Arnold) Brodo & D. Hawksw. (Garrido-Benavent et al. Reference Garrido-Benavent, Pérez-Ortega, De Los Ríos, Mayrhofer and Fernández-Mendoza2021), it cannot be ruled out that comparison of the Galerina sequences along their entire length would reveal some genetic differences.
To the best of our knowledge, the present work is the first examining the temporal origins of Antarctic macrofungal populations. Thus, the Antarctic lineages of Galerina probably diverged from their Northern Hemisphere relatives during the Pleistocene, based on a consensus estimate of divergence times inferred with the various dating strategies implemented in the present study. In G. marginata, the divergence was probably linked to a demographic expansion, as revealed by the calculated neutrality tests. Moreover, the calculated divergence time intervals for the three Galerina agree with the inferred Pleistocene origin of Antarctic populations of amphitropical lichens (Fernández-Mendoza & Printzen Reference Fernández-Mendoza and Printzen2013, Garrido-Benavent et al. Reference Garrido-Benavent, Pérez-Ortega, De Los Ríos, Mayrhofer and Fernández-Mendoza2021). In the lichenized fungi Cetraria aculeata (Schreb.) Fr. and P. minuscula, a close genetic affinity of Maritime Antarctica specimens to South American (Chilean) collections suggested a colonization route through the Sea of Hoces (Drake Passage). In addition, continental specimens of the second species were genetically close to Svalbard (Northern Hemisphere) specimens, which indicated an independent colonization route. The closest relatives of G. marginata, G. badipes and G. fallax, based on the inferred phylogenies, grew in North America and northern Europe, so that a direct, long-distance dispersal across the tropics and ending in the establishment of Antarctic Galerina populations might be assumed. However, the possibility that these species colonized the Antarctic in a series of stepping-stone movements from other territories in the Southern Hemisphere, for which neither specimen nor sequence data are yet available, must not be ruled out. South America was in fact the region from which the vascular plant D. antarctica is believed to have colonized the Antarctic region, also during the Pleistocene (Fasanella et al. Reference Fasanella, Premoli, Urdampilleta, González and Chiapella2017). It is worth recalling that the studied Galerina species grew in tight association with carpets of this plant as well as mosses, where these fungi behave as saprophytes. Even some common populations of Antarctic mosses had a Pleistocene origin (Pisa et al. Reference Pisa, Biersma, Convey, Patiño, Vanderpoorten, Werner and Ros2014, Biersma et al. Reference Biersma, Jackson, Hyvönen, Koskinen, Linse, Griffiths and Convey2017, Reference Biersma, Jackson, Bracegirdle, Griffiths, Linse and Convey2018). The overlapping temporal frameworks for the origins of these plants and fungi that coexist in the same Antarctic terrestrial communities further support a relatively recent Antarctic colonization of Galerina. The meiotic spores produced by their basidiomata, which in general are ellipsoidal or amygdaliform and < 15 μm in length, constitute the expected mode of dispersal, and either wind currents or migratory birds could be involved in such transoceanic dispersals (Muñoz et al. Reference Muñoz, Felicísimo, Cabezas, Burgaz and Martínez2004, Viana et al. Reference Viana, Santamaría and Figuerola2016). For example, Biersma et al. (Reference Biersma, Jackson, Bracegirdle, Griffiths, Linse and Convey2018) inferred aerial models that indicated local wind patterns as the most probable transfer mechanisms from southern South America to the northern Maritime Antarctic. A greater research effort is needed to corroborate these means of dispersal.
The biogeographical history of the studied Antarctic Galerina has been interpreted on the basis of time trees inferred using a secondary calibration (i.e. nrITS substitution rate). Although this approach would be expected to lead to more inaccurate dating results than phylograms calibrated using fossil data (Schenk Reference Schenk2016), the divergence times calculated for the whole Galerina phylogeny in this study are largely coherent with those reported for the divergence of species in other Basidiomycota genera (e.g. Amanita, Heterobasidion, Russula) that used different calibration strategies and more extensive molecular sequence datasets (Chen et al. Reference Chen, Cui, Zhou, Korhonen and Dai2015, Sánchez-Ramírez et al. Reference Sánchez-Ramírez, Tulloss, Amalfi and Moncalvo2015, Looney et al. Reference Looney, Adamčík and Matheny2020). Furthermore, the diversification events within the three Antarctic Galerina species also agree with the inferred colonization events of D. antarctica and mosses that, together with these fungi, form typical terrestrial habitats in Antarctica.
Acknowledgements
The UTM-CSIC technicians are thanked for their assistance during Antarctic fieldwork. The authors also acknowledge Esther Rodríguez (MNCN) for technical assistance in the laboratory; Alexandra Isern, Director Antarctic Earth Sciences, US National Science Foundation, Carolyn Lipke and other personnel at Palmer Station, Polar Programs; the US National Science Foundation for their help collecting Galerina samples from the Antarctic Peninsula; and Benjamin Held, University of Minnesota, for laboratory assistance. This research was part of POLARCSIC activities. Two anonymous peer reviewers are thanked for their feedback.
Funding
This work was supported by grants CTM2017-84441-R (MINECO/FEDER, UE) and PID2019-105469RB-C22 (MICINN, AEI).
Author contributions
IG-B: conceived the project, conducted the analyses and wrote the first draft of the manuscript. RAB: contributed to the molecular dataset and editing of the final manuscript. AdlR: obtained funding and field resources, conducted fieldwork (including specimen collection) and contributed to the editing of the final manuscript.
Supplemental material
A supplemental table will be found at https://doi.org/10.1017/S0954102023000196.







 Open access
Open access