

Creutzfeldt–Jakob disease (CJD) is a devastating illness that is rare and notorious in equal measures. In 1996 a ‘new variant’ (vCJD) was identified (Reference Will, Ironside and ZeilderWill et al, 1996), which is likely to be caused by humans eating beef infected with bovine spongiform encephalopathy (BSE). Although the number of new cases of vCJD has not started rising, the long incubation period means that it will be many years before a major epidemic in humans can be ruled out. In the meantime, representatives of patients with vCJD have expressed concerns about the care that sufferers receive. In particular, patients often present with psychiatric symptoms, but there is some delay before the correct diagnosis is made.
Clinical presentation
Sporadic CJD is rare: there are about 50 new cases a year in the UK, with a peak incidence between 50 and 70 years of age. It is characterised by a rapidly progressive dementing illness, often with severe cerebellar and extrapyramidal signs and myoclonus, causing death usually within a few months (Reference Will and MatthewsWill & Matthews, 1984). Psychiatric symptoms occur early in the disease in about a third of cases (Box 1).
Variant CJD appears to differ from CJD in a number of ways (Reference Will, Ironside and ZeilderWill et al, 1996). For the past 4 years between 10 and 15 new cases have been identified each year (Table 1). Zeidler et al (1997) reported on a series of 14 cases of histologically confirmed vCJD. Onset is usually at a young age, with a mean age of onset of around 30 years. The course of the illness is relatively prolonged, with half the cases surviving more than 1 year. Patients tend to present first to psychiatrists, with anxiety, depression, withdrawal and other behavioural changes. Indeed, in Zeidler et al's series all the patients had early psychiatric symptoms, most commonly depression (Table 2). Four of Will et al's (1996) original series of ten patients had dysaesthesia in limbs or face as the first evidence of neurological involvement. This was generally followed, after a few weeks to months, by gait and limb ataxia. Cognitive impairment appeared relatively late.
History and examination
The early psychiatric symptoms listed in Table 2 are augmented during the course of the illness – apathy/withdrawal, delusions and anxiety are particularly common (occurring in over 85% of cases in a series of 14; Zeidler et al, 1997), and the delusions are often fleeting. Aggression, insomnia, depression and visual and auditory hallucinations may also occur, with first-rank symptoms being present more rarely (in 2 of the 14 cases reported by Zeidler et al, 1997). Subsequently, neurological symptoms become more prominent (Box 2), and paraesthesia or dysaesthesia are common early features of this stage. Particular concern would be raised in a younger person presenting with depression, anxiety or behaviour change who developed neurological symptoms. Diagnosis becomes more obvious as progressive ataxia, involuntary movements and cognitive impairment develop weeks or months after the psychiatric symptoms. A full physical examination will include a neurological examination. Particular neurological signs include cerebellar signs and myoclonus.
Investigations
The investigations of CJD are shown in Box 3. Most cases of sporadic CJD and all cases of vCJD have been homozygous for methionine at codon 129 of the prion protein gene (as are 40% of the normal population). Electroencephalograms often show characteristic periodic triphasic complexes in CJD that are absent in vCJD. Computed tomography and magnetic resonance imaging (MRI) scans are usually normal or show atrophy; MRI may also show symmetrical high signal in the basal ganglia in sporadic CJD, but not in vCJD. However, high signal in the posterior thalamus may prove a useful investigation for vCJD (Reference Zeidler, Sellar and CollieZeidler et al, 2000). In sporadic CJD, cerebrospinal fluid is usually positive for the brain protein 14–3–3 (which may be a marker for neuronal damage), but this test is not useful for vCJD (Reference Zeidler, Stewart and BarracloughZeidler et al, 1997). Tonsillar biopsy has recently been shown to be useful for diagnosing vCJD and it reduces the need for high-risk brain biopsies (Reference Hill, Butterworth and JoinerHill et al, 1999). Researchers in the USA claim to have developed a blood test for prion diseases that is being evaluated ( British Medical Journal, 1999).
Management
If you suspect that a patient has CJD you should contact the UK CJD Surveillance Unit at the Western General Hospital in Edinburgh for advice. Their website (www.cjd.ed.ac.uk) includes information about managing CJD, a short booklet for carers and addresses of support organisations, including the CJD Support Network and the Human BSE Foundation. In addition, a paper is available at the site (www.cjd.ed.ac.uk/carerep.html) that details the experiences of families of 19 patients with CJD. This study identifies a number of concerns of family members. In particular, they were often distressed that their relatives were given a psychiatric diagnosis and that coordination between psychiatric and neurological services was poor. The paper concludes that all patients with a diagnosis of CJD should have a keyworker allocated to coordinate patient and family care. The keyworker should be aware of local services, offer sufficient time and be able to arrange an adequate package of care.
Most patients with CJD require management by psychiatrists and neurologists. Psychiatrists may play a role in educating patients and relatives about the consequences of the disease. Family members and staff are frequently upset by their experiences of nursing a patient with CJD and may seek counselling. Psychiatrists may be asked to give advice about the management of psychiatric symptoms. There are no specific studies of the management of psychiatric illness in CJD. Consequently, management is guided by general principles. Various psychotropic drugs were prescribed to patients with vCJD in Zeidler et al's (1997) study. There was generally little benefit, although three patients showed transient improvement and one appeared to achieve complete, if short-lived, recovery. Anxiety, agitation, restlessness or aggression may be treated with a benzodiazepine such as diazepam, starting with a small dose and gradually titrating against clinical effect. Alternatively, neuroleptics such as risperidone could be prescribed in a similar manner. Antidepressants might be effective for depressive symptoms.
There are at present no treatments for the disease process itself. Various medications have been given without success, including aciclovir, other antivirals and antibiotics. Steroids are often prescribed in the hope that the patient has a hidden cerebral vasculitis. Other symptomatic treatments include clonazepam or sodium valproate for myoclonus, which relatives often find distressing, and pain killers. Patients may become both physically and mentally very unwell, and this can make it difficult to manage them on either medical or psychiatric wards.
Mechanism of infection
Prion diseases occur when prion protein (PrP), a normal brain protein, changes into an insoluble form (Reference Prusiner and HsiaoPrusiner & Hsiao, 1994), largely owing to a change in its shape. The insoluble form is poorly metabolised, accumulates and causes cell death. It can lead to spongiform changes and be deposited as amyloid. The normal soluble form of the protein is PrPC (prion protein, cellular) and its function is unknown. Animals genetically engineered to lack prion protein seem entirely normal (Reference Bueler, Fischer and LangBueler et al, 1992), although it is possible to identify subtle changes in vitro (Reference Collinge, Whittington and SidleCollinge et al, 1994). The insoluble protein was first identified in sheep with the disease scrapie, and is consequently called PrPSc (prion protein, scrapie). This form is resistant to proteases and many of the agents that inactivate proteins, including moderately high temperatures. Once the initial change is triggered, the abnormal protein induces a cascade of conformational change in adjacent normal proteins, so they in turn become insoluble. The initial change from PrPC to PrPSc may occur in different ways (Reference Cohen, Pan and HuangCohen et al, 1994) and is responsible for three different types of prion disease.
Sporadic
In sporadic prion disease, PrP changes to the insoluble form spontaneously. Sporadic CJD currently accounts for over 80% of cases of CJD in the UK.
Inherited
An abnormality in the gene coding for PrP (the prion protein gene) produces a protein that is unstable and changes into PrPSc. Genetic abnormality accounts for about 5% of cases of human CJD. In these cases, the disease displays an autosomal dominant pattern of transmission. Different mutations in the gene can produce different variants of human prion disease. For example, Gerstmann–Straussler syndrome (GSS) results from a point mutation on the prion protein gene and usually presents with cerebellar ataxia (Reference Hsiao, Baker and CrowHsiao et al, 1989).
Transmitted
It was the search for the transmissible agent for diseases like kuru that led Prusiner (1982) to coin the term prion (standing for proteinaceous infectious agent). Prion disease can be experimentally transmitted by directly inoculating the brain with PrPSc. This is the quickest and most reliable route, although incubation takes months and occasionally years. In humans, iatrogenic transmission of CJD has occurred through contaminated neurosurgical instruments and the use of cadaveric pituitary extracts. Iatrogenic CJD currently accounts for about 5% of CJD in the UK. Infection can also follow eating PrPSc, although transmission is generally more difficult. Kuru was maintained through cannibalistic rituals, demonstrating that the oral route can lead to disease in humans. The incubation time for kuru can be as long as 20 to 30 years (Reference Prusiner, Gajdusek and AlpersPrusiner et al, 1982).
Spread across species
Although all animals have some form of prion protein, the types vary between species, as does the ease with which it can be transmitted from one species to another. For example, it appears that scrapie in sheep, which has been endemic in many sheep-rearing areas for hundreds of years, cannot directly cause disease in humans (Reference WillWill, 1993).
In experimental conditions, the GSS prion protein can induce disease in monkeys and rodents, and the BSE prion protein can induce disease in mice, sheep, goats and monkeys. The BSE epidemic that occurred in UK cattle may have been caused by exposure of cattle to sheep prion in animal feed.
Although there are different forms of prion in different species, the clinical picture of prion disease across species is similar: a rapidly progressive and fatal degeneration of the central nervous system, often with prominent ataxia.
Pathology
The pathology shows spongiform change that consists of fine vacuolation of the neuropil of the grey matter. This is associated with astrocytosis and neuronal loss. Amyloid plaques, although common in the other human spongiform encephalopathies, kuru and GSS, are observed in only 5–10% of patients with CJD (Reference Bell and IronsideBell & Ironside, 1993), but are more common in vCJD. All of these conditions are called spongiform encephalopathies and all show similar spongiform change in the brain.
Implications of the disease process
Study of CJD and the other prion diseases has drawn attention to the potential importance of abnormal protein folding as a pathogenic mechanism for central nervous system diseases. First, a misfolded protein may induce conformation change in adjacent normally folded proteins; a cascade results in the formation of large quantities of misfolded protein in a previously healthy brain. Second, whereas the normally folded protein is soluble, the misfolded protein is insoluble and forms aggregates that are deposited in or around the cell. The result is cell death. Very similar processes may be taking place in Alzheimer's disease, with the deposition of amyloid, and Huntington's disease, in which insoluble huntingtin protein accumulates. In each case an altered protein conformation may act as the seed for further misfolded proteins and a subsequent cascade.
It may be possible to inhibit the pathogenic cascade in these neurodegenerative diseases by making use of the ‘molecular chaperones’, also called heat-shock proteins, involved in the various steps of protein folding (Reference Satyal, Schmidt and KitagawaSatyal et al, 2000). These molecular chaperones are induced by the presence of a misfolded protein, and their role may be to inhibit its potentially lethal effects.
Public health issues
There is evidence to suggest that people who develop vCJD were exposed to BSE, and since the incubation period may be long, there remains the possibility of a human epidemic (Reference CollingeCollinge, 1999). BSE was first formally identified in cattle in the UK in 1986. It resembles scrapie and is thought to have been spread by the practice of feeding sheep offal to cattle in the 1970s and 1980s. The practice was banned in 1988; possibly contaminated offal was removed from the food chain the following year. Reports of the first cases of vCJD in humans were published in 1996 (Reference Will, Ironside and ZeilderWill et al, 1996), and the first reliable suggestions of a link with BSE emerged in 1997. Strenuous efforts, including the mass incineration of cattle, have been made to remove BSE from the food chain. The number of BSE cases in cattle in the UK is expected to decline from over 2000 in 1999 to fewer than 500 in 2001.
Monitoring new cases
The UK CJD Surveillance Unit in Edinburgh monitors the number of cases of CJD and publishes the results on the internet (Table 1). There is now also a large screening programme to examine removed tonsils and appendixes for PrPSc. Initial results from studies of over 3000 appendixes were encouraging (none was positive), but the sample is presently too small to exclude an epidemic and the series is being increased (Reference Ironside, Hilton and GhaniIronside et al, 2000).
Recent research from Oxford suggests that a major epidemic is less likely than previously thought (Reference Ghani, Ferguson and DonnellyGhani et al, 2000). Modelling the latest data shows that no more than two cases of vCJD could arise from the consumption of a single cow. Given that thousands of people may eat material from a single cow, this implies a substantial barrier.
Spread in humans
Since the number of people incubating this disease is unknown, there are concerns that it might be spread via blood, tissue donation or surgical instruments.
At present, it is not possible to screen blood for CJD, and concerns about contamination have led the US Food and Drug Administration to ban blood donations from anyone who has spent more than 6 months in the UK between 1980 and 1997 (when exposure to CJD in the UK was considered to be at its peak). As B-lymphocytes have been shown to be required for infection to occur by blood transfusion in mice, blood in the UK is now leucodepleted, a process that removes up to 95% of white blood cells (Reference CollingeCollinge, 1999).
Transmission via contaminated surgical instruments is possible, because normal hospital sterilisation is unlikely to inactivate prions completely. When instruments are likely to be contaminated (for example, in a tonsil biopsy from someone with suspected CJD) they are not reused. The risk of transmission during routine surgery is not known. To eliminate completely the risk of spread, all surgical instruments would have to be disposable. However, the cost of this would be impractical. One suggestion is to use a recording system for all operations, to allow instruments used on patients who later develop vCJD to be traced and destroyed.
Summary
Creutzfeldt–Jakob disease is a devastating illness that is unusual from at least three perspectives: the prion disease process (prion diseases are unique in being both heritable and transmissible), the clinical presentation, often accompanied by psychiatric symptoms, and the public health implications. Unravelling the disease process may tell us more about the pathogenesis of other dementias. CJD is a rare condition and, we must hope, will remain so. Although at the present rate the average consultant psychiatrist should expect to see a case only once every 18 years, earlier diagnosis may be achieved by being alert to the condition and its presentation. Relatives are anxious to receive well-resourced and coordinated care.
Multiple choice questions
-
1. Regarding sporadic CJD:
-
a there is about 1 new case per consultant psychiatrist per year in the UK
-
b the mean age of onset is 30 years
-
c psychiatric symptoms occur in about 10% of cases
-
d dementia occurs early in the illness
-
e tonsil biopsies are positive for prion protein.
-
-
2. Regarding variant CJD:
-
a there are 10–15 new cases each year in the UK
-
b the mean age of onset is 60 years
-
c about two-thirds of patients present with psychiatric symptoms
-
d CT scan is usually normal
-
e death occurs after a mean of 14 months.
-
-
3. Presenting symptoms of vCJD include:
-
a depression
-
b withdrawal
-
c agitation
-
d delusions
-
e insomnia.
-
-
4. Concerning the management of CJD or vCJD:
-
a psychiatrists are normally consulted early in the illness
-
b relatives generally feel that psychiatrists and neurologists work well together
-
c a keyworker is not helpful for such a serious illness
-
d aciclovir or other antivirals are sometimes helpful
-
e as yet no useful information is available on the internet.
-
-
5. Regarding public health issues:
-
a the number of new cases of vCJD is increasing each year
-
b the UK is as likely to have an epidemic of vCJD as the USA
-
c vCJD can be transmitted by sterilised surgical instruments
-
d blood is now routinely screened for CJD
-
e 40% of the population are at a higher risk of developing vCJD.
-
Table 1 Deaths resulting from Creutzfeldt–Jakob disease (CJD) and definite and probable variant CJD (vCJD) in the UK to 6 November 2000 (data from UK CJD Surveillance Unit)
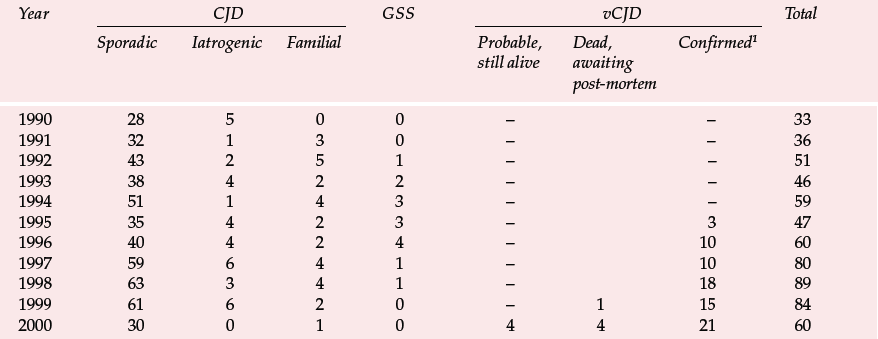
| Year | CJD | GSS | vCJD | Total | ||||
|---|---|---|---|---|---|---|---|---|
| Sporadic | Iatrogenic | Familial | Probable, still alive | Dead, awaiting post-mortem | Confirmed1 | |||
| 1990 | 28 | 5 | 0 | 0 | – | – | 33 | |
| 1991 | 32 | 1 | 3 | 0 | – | – | 36 | |
| 1992 | 43 | 2 | 5 | 1 | – | – | 51 | |
| 1993 | 38 | 4 | 2 | 2 | – | – | 46 | |
| 1994 | 51 | 1 | 4 | 3 | – | – | 59 | |
| 1995 | 35 | 4 | 2 | 3 | – | 3 | 47 | |
| 1996 | 40 | 4 | 2 | 4 | – | 10 | 60 | |
| 1997 | 59 | 6 | 4 | 1 | – | 10 | 80 | |
| 1998 | 63 | 3 | 4 | 1 | – | 18 | 89 | |
| 1999 | 61 | 6 | 2 | 0 | – | 1 | 15 | 84 |
| 2000 | 30 | 0 | 1 | 0 | 4 | 4 | 21 | 60 |
Table 2 Presenting features and psychiatric diagnosis in 14 cases of variant Creutzfeldt–Jakob disease (vCJD) (Zeidler et al, 1997, with permission)
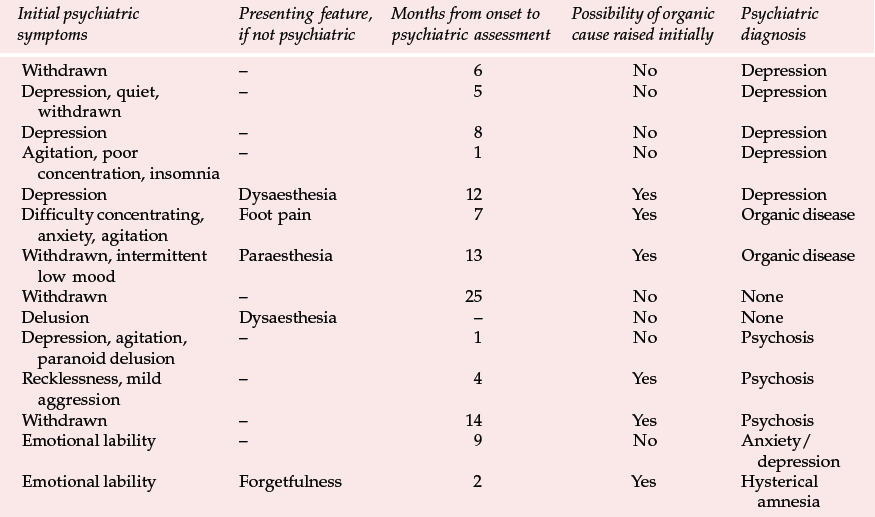
| Initial psychiatric symptoms | Presenting feature, if not psychiatric | Months from onset to psychiatric assessment | Possibility of organic cause raised initially | Psychiatric diagnosis |
|---|---|---|---|---|
| Withdrawn | – | 6 | No | Depression |
| Depression, quiet, | – | 5 | No | Depression |
MCQ answers
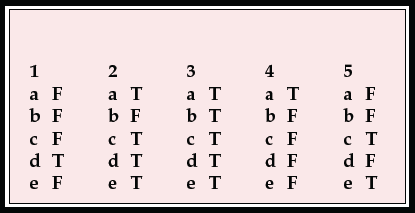
| 1 | 2 | 3 | 4 | 5 | |||||
| a | F | a | T | a | T | a | T | a | F |
| b | F | b | F | b | T | b | F | b | F |
| c | F | c | T | c | T | c | F | c | T |
| d | T | d | T | d | T | d | F | d | F |
| e | F | e | T | e | T | e | F | e | T |



eLetters
No eLetters have been published for this article.