


Summations
-
∙ This review summarises the evidence that chronic low grade inflammation contributes to the pathophysiological changes associated with major depressive disorder (MDD). Such information might be of value in the development of a new generation of antidepressants.
-
∙ There is evidence from clinical and experimental studies to support the hypothesis that chronic inflammation not only contributes to the structural and functional changes in the brain but is also important in causing physical ill health (heart disease, diabetes, cancer, etc.) which occurs frequently in MDD patients.
-
∙ Evidence is presented that the neurodegeneration attributed to inflammation contributes
-
to the onset of dementia in a proportion of elderly patients.
-
∙ The review concludes by summarising the possible development of novel antidepressants based on drugs that reduce the impact of low grade inflammation. Preliminary therapeutic evidence suggests that anti-inflammatory drugs which reduce prostaglandin E2 might indicate how non-monoamine modulating antidepressants could be developed.
Considerations
-
∙ The review is not based on an exhaustive and fully comprehensive assessment of all the recent publications on the immune changes reported to occur in patients with MDD. Thus many of the studies cited, although they have been subjected to appropriate statistical analysis, are based on ‘single point’ data from relatively small numbers of patients.
-
∙ In most studies cited it is difficult to distinguish between the biological significance and the statistical significance of the changes reported. For example, the increase in the serum concentration of the proinflammatory cytokines may be only 10% greater than the control concentration. Is this alone sufficient to initiate the dramatic changes reported in MDD?
Introduction
It is somewhat surprising to find that only relatively recently has the topic of neuroinflammation appeared in most textbooks of psychiatry, psychopharmacology and neuroscience. Indeed, the topic of inflammation was previously restricted to the domain of immunology and infectious diseases as the brain was considered to be an immune privileged organ and therefore largely unaffected by immune changes in the periphery. Thus, unless the blood–brain barrier (BBB) was damaged by infection or trauma, it was widely accepted that immune-related changes could largely be ignored by psychiatrists and psychopharmacologists.
The situation changed dramatically some 20 years ago with the discovery that that the immune system could profoundly affect the mood state independently of the somatic changes due to an acute infection. The subsequent discovery that proinflammatory cytokines played a crucial role in the generation of the psychic symptoms added further to the developing specialty of psychoneuroimmunology.
This article will give an overview of the importance of chronic inflammation in the aetiology of depression and illustrate how such knowledge may open up new possibilities for novel drug development.
Inflammation, the immune system and the link to the pathophysiology of depression
The immune communication between the brain and the periphery is bi-directional. Whereby afferent pathways are connected to the brain by cytokines, sensory fibres and the vagus nerve while the efferent pathways modulate the peripheral immune responses via the hypothalamic–pituitary–adrenal (HPA) axis, the sympathetic and the parasympathetic systems. Thus both neuronal and humoral processes are involved in the coordination of the physiological, immunological and behavioural responses following and inflammatory challenge (Reference Quan and Banks1) (Fig. 1).
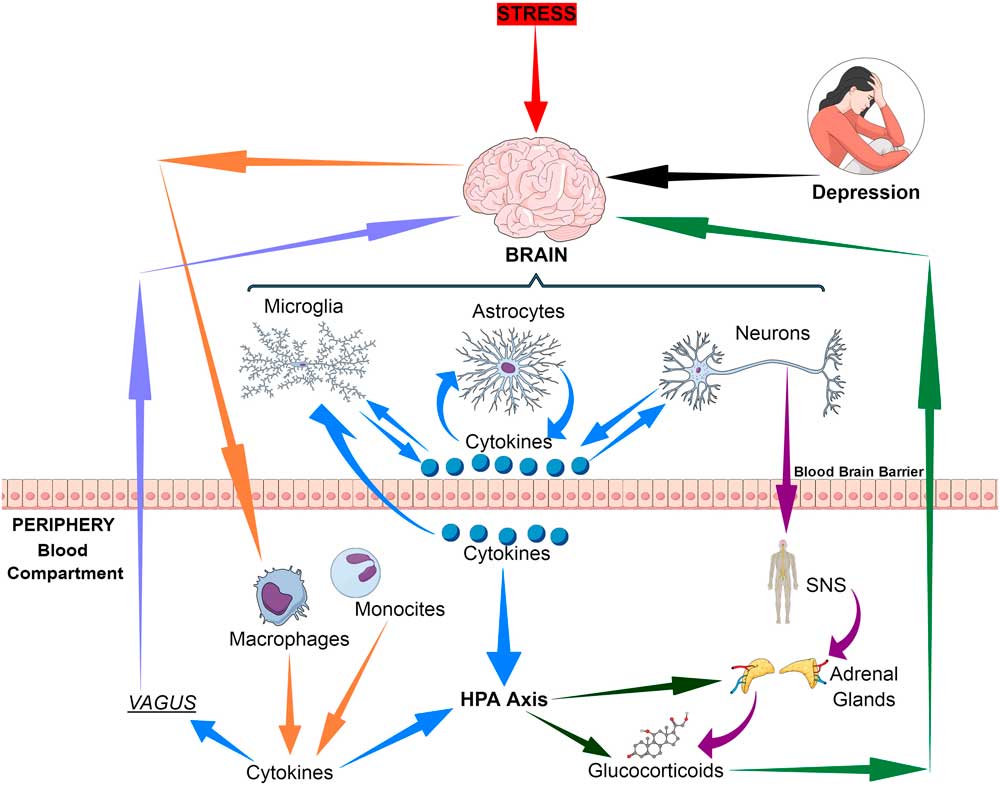
Fig. 1 Bio-directional connections between the stress, the brain and inflammatory cytokines. HPA, hypothalamic–pituitary–adrenal axis; SNS, sympathetic nervous system.
Inflammation is usually a reflection of cell damage caused by infections, physical injury or the response of tissues to an antibody challenge. However, in recent years it has become apparent that psychological stress can also initiate the inflammatory response, thereby linking inflammation to both physical and mental ill health.
Following an infection, the ability to mount an inflammatory response is an essential part of survival but the inflammatory response may occasionally be exaggerated and over prolonged thereby leading to adverse effects, for example in chronic arthritis or diabetes.
But besides the classical inflammatory response to infection, the immune changes associated with major mental disorders are not characterised by the classical inflammatory signs of colour (warmth), delor (pain) and tumour (swelling). The term ‘neuroinflammation’ is used to describe the immune-related process which occurs within the brain and spinal cord and is a result of the injury caused by infection, psychological or physical stress, or indirectly as a consequence of infection arising in the periphery. As a consequence of neuroinflammation, the innate immune cells in the brain (the microglia, astrocytes and oligodendroglia) are activated to liberate cytokines, chemokines and secondary inflammatory mediators, such as the prostaglandins, in response to the inflammatory stimulus.
Whereas an acute infection of the central nervous system (CNS) is usually beneficial and neuroprotective, chronic inflammation is usually maladaptive and has a detrimental effect on brain homoeostasis. This is particularly apparent in major neurological disease such as multiple sclerosis, stroke, Alzheimer’s disease or Huntington’s disease (Reference Lee, Han, Nam, Oh and Hong2). However, there are important differences between these neurological disorders and the neuroinflammation associated with non-pathological conditions such as chronic stress. Such conditions do not usually involve a major disruption of the BBB but are primarily due to the activation of localised immune mechanisms resulting in the activation of the central sympathetic system, the HPA axis and proinflammatory mediators. Such changes are often associated with sickness behaviour, characterised by fatigue, anorexia, anhedonia, sleep disturbance and loss of libido. This will be discussed later in the review.
Hitherto the brain has been considered to be an immune privileged organ. The immune response is divided into the innate and adaptive components. This the innate system reacts to pathogens by recognising their specific pathogen-associated molecular patterns which initiates the release of histamine, prostaglandins, bradykinin, serotonin and leucotrienes. These inflammatory factors may produce a localised inflammatory response, such as vasodilatation and pain, but also attract macrophages to the infection site which release such proinflammatory cytokines as tumour necrosis factor α (TNF), interleukins (IL-) 1 and 6. The cytokines then prolong the systemic inflammatory response and attract other immune cells, leucocytes and lymphocytes, to the inflammatory site.
The adaptive immune response functions to produce a memory of the immune events. T-cells produce the cellular response that has a toxic effect on potentially harmful microorganisms, cancer cells, etc., and they maintain the immune response by releasing cytokines such as IL-2 that attract macrophages, neutrophils and leucocytes/lymphocytes. The B-lymphocytes induce the humoral response in which antibodies are produced against the specific pathogen. For a detailed account of the structure of the immune system in health and disease (Reference Abbas, Lichtman and Pillai3).
The brain lacks the adaptive arm of the immune system, lacking the T- and B-cells that are involved in cellular and humoral immunity. However, the T-cells from the periphery do survey the CNS in the meninges, choroid plexus and cerebrospinal fluid (CSF) (Reference Hickey4). Normally the BBB acts as a protection for the brain by limiting access from the periphery of large molecules, cells and microorganisms but it is important to stress that the BBB is not just a physical barrier but also a means whereby neuroimmune communication can occur between the brain and the periphery. However, the selectivity of the BBB for the passage of large molecules and cells depends on the region of the brain, the choroid plexus and the circumventricular region being more permeable to large molecules such as neuropeptides than other regions (Reference Ermisch, Ruhle, Landgraf and Hess5).
The BBB consists of endothelial cells, pericytes and neuroglia. The endothelial cells form tight junctions due to the junctional proteins, the occludins and claudins, which limit the penetration by large molecules such as the cytokines, The stability of the BBB is further enhanced by the astrocytes and pericytes that cover the vasculature. Peripheral immune cells become attached to endothelial cells by means of the intracellular adhesion molecules such as ICAM-1, adhesion being enhanced by the action of IL-1β. There is also evidence that peripheral immune cells can enter the CNS due to the degredation of the astrocyte-pericyte sheath thereby enhancing neuroinflammation (Reference Greenwood, Wang and Calder6).
IL-1β is activated from its biologically inactive precursor by adenosine triphosphate (ATP). ATP then activates P2×7 purinergic receptors and the NLRP3 inflammasome complex that, in combination with caspase-1 cleaves pro-IL-1β (Reference Ferrari, Pizzirani and Adinolfi7). In the brain, the inflammosone complex is expressed in microglia located in the hippocampus and other mood regulating regions that are particularly vulnerable to the effects of chronic stress (Reference Iwata, Ota and Duman8). This mechanism helps to explain how proinflammatory cytokines can exacerbate the effects of stress on brain function in depression.
The microglia are the primary immune cells in the brain and exist in two phenotypic forms. Under normal conditions, microglia have a non-immune function such as the removal of dead neurons, clearing aberrant synapses during neurodevelopment and monitoring synaptic structures. The microglia are also involved in patrolling the microenvironment and removing invading pathogens.
This is achieved by the production of pseudopodia and thereby resembling the macrophages of the periphery (Reference Nimmerjahn, Kirchhoff and Helmchen9). The detection of pathogens by the microglia is facilitated by Toll-like receptors (TLR’s). In addition, the microglia also contain surface membrane receptors for neurotransmitters and hormones. The microglia are functionally similar to the peripheral macrophages by being activated by antigen presenting cells via the TLR’s. In addition, they produce both inflammatory (TL-1β, IL-6, TNF-α) and anti-inflammatory (IL-10, transforming growth factor (TGF)-β) cytokines. The activated microglia also initiate a cascade of chemokines, prostaglandins and nitric oxide (NO). In addition to their direct contribution to inflammation in the brain, chemokines such as CCR2 enhance the recruitment of peripheral immune cells into the brain and thereby increase the impact of inflammation (Reference Prinz and Priller10).
While the activated microglia are clearly beneficial in protecting the brain from infection, the prolonged neuroinflammatory changes may be detrimental. Such a process probably occurs in the ageing brain, in chronic psychiatric disorders such as depression and schizophrenia and particularly in the neurodegenerative diseases. These mechanistic aspects have been the subject of a series of experimental studies by Godbout et al. (Reference Godbout, Moreau and Lestage11) and by Wynne et al. (Reference Wynne, Henry, Huang, Cleland and Godbout12). Such mechanisms could form the basis for the neurodegenerative changes arising in chronic depression and thereby act as a prelude to dementia in old age (Reference Leonard13).
In addition to the microglia, the astrocytes also play a vital role in the immune system in the brain. Previously the astrocytes were envisaged as providing structural support for neurons but it is now evident that they maintain the integrity of the BBB, regulate synaptic neurotransmitter function and remove neurotoxins that are formed as end products of the tryptophan–kynurenine pathway. A further important function of the astrocytes is in the control of the local glutamate concentration which, if present in excess, causes apoptosis of neurons by prolonged activation of the N-methyl-d-aspartate (NMDA) receptors. In this way, the astrocytes control the excitotoxic potential of glutamate by converting it into glutamine and shunting glutamine into neurons where it is re-synthetised into glutamate (Reference Galic, Riazi and Pittman14). The activation of the astrocytes has been shown to occur both in schizophrenia and major depression where they are presumed to have a neuroprotective role (Reference Rothermundt, Falkai and Ponath15).
The cytokines play a central role in regulating the immune system and integrating it with the endocrine system and with neurotransmitter function. This role is achieved due to the presence of cytokine receptors on immune cells, neurons and the main components of the HPA axis; neurotransmitter and endocrine receptors also occur on macrophages, microglia and astrocytes.
Activation of peripheral and central macrophages results in the release of pro- and anti-inflammatory (IL-1β, IL-6, TNF-α, IFN-γ and IL-antagonist and TGF-β, respectively). This mechanism involves the activation of nuclear factor kappa-beta (NF-kB) (Reference Parker, Luheshi, Rothwell and Pinteaux16). In addition, the cytokines promote the synthesis of other cytokines, chemokines and prostaglandins (PGE2) which also activate the HPA axis (Reference Ericsson, Arias and Sawchenko17).
Another important mediator of neuroinflammation is the gaseous neurotransmitter NO, the synthesis of which is increased due to the induction of nitric oxide synthase (iNOS) by inflammatory cytokines. Whereas under non-inflammatory conditions the concentration of NO is low and acts and acts as a gaseous transmitter at glutamatergic neurons, high concentrations of NO generated by neuroinflammation cause neuronal damage by activating glutamatergic neurons and causing the nitrosation of nucleic acids (Reference Paakkari and Lindsberg18) (Fig. 2).
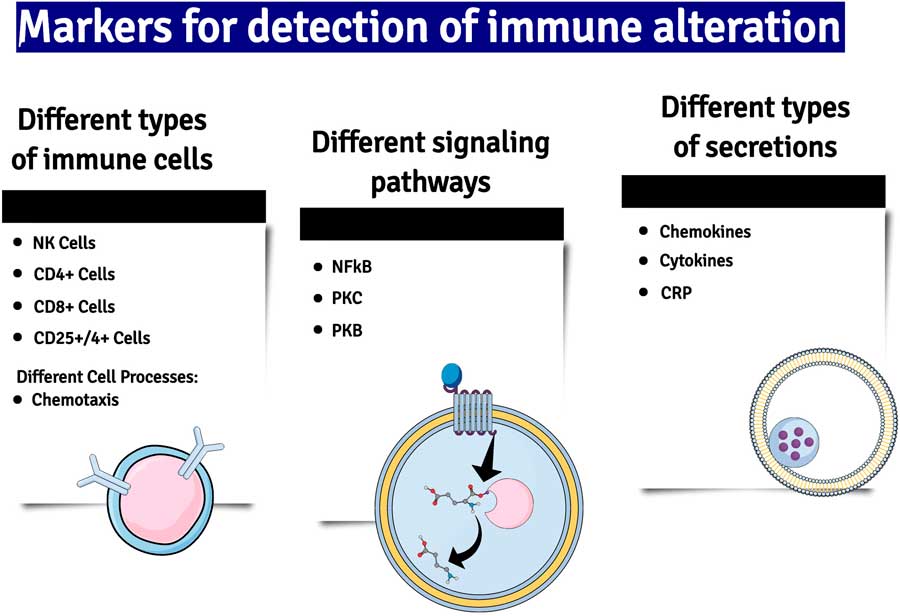
Fig. 2 A summary of some of the major changes in inflammatory markers which have been detected in patients with major depression. NK cells, natural killer cells; NF-kB, nuclear factor kappa B; PKC and B, phosphokinese C and B; CRP, C-reactive protein.
The effect of inflammation on the endocrine-immune systems
Stress-induced hypercortisolaemia forms part of the adaptive mechanism in the ‘fight or flight’ response. However, if the stress response is prolonged and becomes dissociated from the initiating stimulus it becomes detrimental to the brain and the peripheral organs. For example, in patients with major depression hyperactivity of the HPA axis, as reflected in hypercortisolaemia, occurs in ~50% of patients but increases to about 80% in those who are severely depressed (Reference Pariante and Miller19). These changes are reflected by the increase in cortisol in the blood, CSF and urine together with an enlargement of the adrenal and pituitary glands (Reference Juruena, Cleare, Papadopoulos, Poon, Lightman and Pariante20).
However, despite the presence of increased glucocorticoids which would usually be associated with a reduction in proinflammatory mediators, major depression is frequently associated with an increase in proinflammatory cytokines, chemokines and prostaglandins (Reference Pollak and Yirmiya21,Reference Raison, Capuron and Miller22).
Stress, whether psychological or physical, is an important trigger for the onset of most major psychiatric disorders. However, stress-induced hypercortisolaemia is a component of the adaptive mechanism in the ‘fight or flight’ response as elucidated by Cannon in the 1930s and it is only if the stress response is prolonged, and is dissociated from the initiating stimulus, that it becomes detrimental to the brain and peripheral organs.
Clinical and experimental studies have shown that the nature of the change in the HPA axis, and its impact on the immune system, occurs with the duration of the stress stimulus (Reference Segerstrom and Miller23), the effects of acute stress on the immune system differing from chronic stress. The changes linking chronic stress with cellular immunity involve changes in the expression and sensitivity of glucocorticoid receptors (GRs) (Reference Quan, Avitsur and Stark24). In this respect, proinflamatory cytokines have been shown to increase the inactive α GR isoform and decrease the active β form of the receptors (Reference Pace and Miller25). Such changes are termed glucocorticoid receptor desensitisation and thereby reflect the decrease response to the circulating glucocorticoids.
The quantitative and qualitative effects of major depression differ from those changes in the endocrine-immune system seen following an acute stress response. A major difference relates to the neurodegenerative changes associated with chronic inflammation. The glucocorticoids affect the integrity of neuronal membranes by blocking the neuronal repair mechanisms that are carried out by neurotrophic factors such as brain-derived neurotrophic factor (BDNF). BDNF, and other neurotrophic factors, are peptides and peptide synthesis in the brain and periphery is reduced by the glucocorticoids. This results in a reduction in the repair of damaged dendrites, neurites and axons. This effect of the glucocorticoids has been shown both experimentally and clinically (Reference Lucassen, Meerlo and Naylor26). There is also evidence that some proinflammatory cytokines such as IL-1β reduce hippocampal neurogenesis (Reference Zunszain, Anacker and Cattaneo27,Reference Goshen, Kreisel and Ben-Menachem-Zidon28).
Following the acute stress response, glucocorticoids suppress the expression of inflammation but due to GR desensitisation following chronic stress, this mechanism is suppressed thereby resulting in an increase in the inflammatory state. This situation is enhanced by the chronic activation of the HPA axis and leads to an increased release of glucocorticoids combined with decrease in the plasma binding capacity of corticosterone binding globulin (Reference Stefanski29). These changes are associated with a cellular reduction in the translocation of GRs and a reduction in the reactivity of GR protein in immune cells (Reference Quan, Avitsur and Stark30). The reduced expression of the GRs occurs in peripheral tissues and also in the hippocampus (Reference Quan, He, Lai, Shen and Herkenham31), a region that is crucially involved in the negative feedback regulation of the HPA axis (Reference De Kloet, Vreugdenhil, Oitzl and Joels32). This mechanism help to explain the apparent paradox why, in patients with chronic depression, there is an elevation of plasma and tissue glucocorticoids coincidentally with a increase in proinflammatory mediators.
Experimental studies have demonstrated that chronic stress changes the microglia from the resting to the active state in a number of brain regions (Reference Sugama, Fujita, Hashimoto and Conti33) and there is limited evidence that this also occurs in depressed patients (Reference Steiner, Bielau and Brisch34). Furthermore, clinical studies have shown that chronic social stress increases peripheral macrophage activity which provides indirect evidence in support of stress-induced microglial activation in depression. (Reference Glaser, Robles, Sheridan, Malarkey and Kiecolt-Glaser35).
The changes in the microglia are accompanied by a reduction in the astrocytes and oligodendroglia in many regions of the brain, including the amygdala and the sub-genual prefrontal cortex (Reference Ongur, Drevets and Price36). The amygdala and prefrontal cortex in particular have been implicated for being dysfunctional in major depression while the reduction in astrocytes could be linked to the neurodegenerative changes which occur as a consequence on the inflammation induced increase in the excitotoxin quinolinic acid (Reference Myint37). This will be discussed in a later section of this review.
Chronic stress also suppresses neuronal repair and neuroregeneration. Duman et al. (Reference Duman, Heninger and Nestler38) provided experimental evidence to show that the stress induced increase in proinflammatory cytokines is associated with a decrease in the synthesis of BDNF, changes that are reversed by chronic antidepressants. Thus the reduction in neuronal repair caused by a reduced synthesis of neurotrophic repair factors, combined with a more generalised decrease in protein synthesis due to hypercorticolaemia, contribute to the changes in brain structure that characterise chronic depression.
It is often overlooked that the glucocorticoids have both pro- and anti-inflammatory actions depending upon their concentration. For example, in experimental studies it has been shown that basal cortisol concentrations exert an anti-inflammatory effect while higher concentrations, equivalent to those seen following a major physical stress, enhance the inflammatory response that is initiated by lipopolysaccharide (LPS) (Reference Yeager, Pioli and Guyre39). This proinflammatory effect is a consequence of the activation of the MAPK signalling and NF-kB pathway which are essential for the synthesis of proinflammatory cytokines and chemokines (Reference Smyth, Stapleton and Freeman40).
An important aspect of the glucocorticoid-inflammation hypothesis of depression relates to the relationship between the concentrations of the glucocorticoids and the proinflammatory cytokines and the symptoms of depression. Many of the clinical and experimental studies quoted to support the hypothesis are based on acute studies in which high doses of synthetic glucocorticoids were administered. However, there is now sufficient evidence that the proinflammatory effects of the glucocorticoids, and the plasma concentration of the proinflammatory cytokines, in patients with major depression approximately coincide with those found to elicit similar changes seen in the experimental studies (Reference Yeager, Pioli and Guyre39,Reference Maes, Bosmans, De Jongh, Kenis, Vandoolaeghe and Neels41).
In addition to the reduction in neuronal repair, neurodegeneration also occurs in the depressed patient brain and the more frequent and severe the depressive episodes the greater the neurodegeneration particularly in the hippocampus, frontal cortex and amygdala (Reference Maes, Yirmyia and Noraberg42,Reference Sheline, Mittler and Mintun43). Such changes are clearly linked to the neurotoxic effects of hypercortisolaemia and proinflammatory cytokines, Additional neurodegenerative changes are associated with the synthesis of the glutamate NMDA agonist, quinolinic acid, together with xanthenuric acid and other intermediates which result in the synthesis of reactive oxygen species. These neurotoxic compounds are products of the tryptophan–kynurenine pathway and the activation of this pathway is due to the action of proinflammatory cytokines on indoleamine dioxygenase and kynurenine monooxygenase (Reference Myint and Kim44) and will be considered in more detail later.
Inflammation, sickness behaviour and depression
Sickness behaviour is a reflection of immune activation and is commonly associated with depression, the chronic fatigue syndrome and certain types of cancer such as pancreatic cancer. Thus fatigue, sleep disturbance, hyperalgesia, anorexia and loss of libido are frequently associated with these conditions and often resemble infectious disease symptoms such as influenza, hence the term ‘sickness behaviour’. However, the question arises whether sickness behaviour seen in psychiatric disorders is part of a continuum that develops into major depression or is separate from the pathological processes which result in depression. Perhaps the evolutionary importance of sickness behaviour provides a clue!
The symptoms that arise in all animal species following a systemic infection are essentially similar. The neurovegetative symptoms include lethargy, anorexia, hypersomnia, loss of libido and, in humans, anhedonia. Such symptoms would be beneficial for survival as they allow the animal to withdrawal into a safe environment so that the healing process may occur. The term ‘sickness behaviour’ was devised to describe such behaviour (Reference Kent, Bluthe, Kelley and Dantzer45). The evolutionary advantage for such behaviour would also occur as a result of the avoidance of contact with others in the group and thereby reduce the spread of the infection. This suggests that sickness behaviour is a relatively short-term reaction to an acute inflammatory challenge and reflects a strategy that is critical for the survival of the individual, a concept first proposed by Hart (Reference Hart46).
However, the situation which occurs when inflammation becomes chronic is likely to be detrimental to survival as occurs, for example, in autoimmune diseases, cardiovascular disease, diabetes and as a consequence of the secondary effects of obesity when mood symptoms often predominate and even worsen the outcome (Reference Raison and Miller47). As chronic low grade inflammation is a characteristic feature of affective disorders, it would seem reasonable to conclude that sickness behaviour is distinct from depression but does serve an important survival function (Reference Janicki-Deverts, Cohen, Doyle, Turner and Treanor48). In support of this view, Janicki-Deverts et al. (Reference Janicki-Deverts, Cohen, Doyle, Turner and Treanor48) assessed the relationship between sickness behaviour and proinflammatory cytokines by assessing the response of group of healthy adults to an influenza rhinovirus challenge. The results showed that the increase in IL-6, IL-1β and TNF-α is associated with a reduced positive affect but not with a negative affect thereby suggesting that there were differences between sickness behaviour, depression and the changes in peripheral proinflammatory cytokines.
As antidepressants are reasonably effective in attenuating the symptoms of depression, are they equally effective in attenuating low grade inflammation which occurs concurrently with the major symptoms? From rodent models of depression there is certainly evidence that antidepressants reduce the tissue concentrations of IL-6 and TNF-α (Reference Yirmiya, Pollak and Barak49–Reference Yaron, Shirazi, Judovich, Levartovsky, Caspi and Yaron51).
However, these conclusions are based on the effects of antidepressants on in vitro or acute in vivo models in which lipopolysaccharide was used as an acute immune challenge. A more representative model of major depression is provided by the olfactory bulbectomised rat model which not only demonstrates the elevation of tissue proinflammatory cytokines, prostaglandin E2 and NO but also shows that chronic treatments attenuate the depression-like symptoms concurrently with the tissue inflammatory mediators (Reference Song and Leonard52).
In contrast to the largely positive results from the chronic rodent studies, it has also been shown that chronically administered fluoxetine increased, rather than decreased, some of the proinflammatory markers (Reference Myint, O’Mahony and Kubera53).
Despite these positive results from rodent studies, those based on changes in the blood concentrations in patients are less convincing. At best, the clinical results are inconsistent and several studies have reported no changes in the cytokines in depressed patients despite an adequate response to treatment (Reference Jazayeri, Keshavarz and Tehrani-Doost54,Reference Maes, Meltzer and Bosmans55). Indeed, there is evidence from experimental studies that chronic fluoxetine treatment increases the concentration of proinflammatory cytokines (Reference Lee, Rao, Ertley, Chang, Rapoport and Bazinet56). Thus, overall, it would appear that antidepressants currently available have an inconsistent effect on inflammation in depression.
Inflammation, neurotoxins and neurodegeneration in chronic depression
Epidemiological studies have reported that the frequency of Alzheimer’s disease and other neurodegenerative disorders increases in those who have chronic depression in comparison to an age and gender matched control population (Reference Geerlings, den Heijer, Koudstaal, Hofman and Breteler57). These observations are supported by the finding that a history of depression is an important risk factor for dementia in later life (Reference Jorm58).
Pathological finding have also shown that the frequency of amyloid plaques and neurofibrillary tangles is also greater in patients with Alzheimer disease who had been diagnosed with major depression (Reference Rapp, Schnaider-Beeri and Grossman59,Reference Sun, Steffens and Au60). As mentioned previously, neurodegenerative changes in the hippocampus, and prefrontal cortex has been reported to occur in patients with major depression (Reference Sheline, Mittler and Mintun43). Thus it would appear that the neurodegenerative changes, and severe cognitive deficits, are a frequent outcome of major depression. It is therefore of importance to consider how chronic inflammation may contribute to such changes. This has been reviewed by Leonard (Reference Leonard61) and Leonard and Myint (Reference Leonard and Myint62).
Over 40 years ago, Lapin et al. published a series of experimental studies demonstrating that depressive-like behaviour was the result of metabolic end products of the tryptophan–kynurenine pathway (Reference Lapin and Oxenkrug63,Reference Lapin64). Of these end products, the authors demonstrated that quinolinic acid and 3-hydroxykynurenine caused anxiety and stress-like changes in rodents, thereby suggesting that there was a neurochemical link between anxiety and depression (Reference Lapin64).
In recent years there has been a renewed interest in the tryptophan–kynurenine pathway which has been stimulated by the reduction in brain serotonin and plasma free tryptophan, and an increase in the metabolites of kynurenine in the blood and CSF in major depression.
Under non-stress conditions tryptophan is primarily metabolised in the liver to kynurenine by tryptophan 2,3-dioxygenase under the influence of circulating glucocorticoids (Reference Satyanarayana and Rao65). However, in stressful and inflammatory conditions tryptophan is metabolised by indoleamine 2,3-dioxygenase (IDO) which is widely distributed in immune cells, lung, kidney and the brain (Reference Carlin, Borden, Sondel and Byrne66,Reference Taylor and Feng67). Anti-inflammatory cytokines reduce the activity of IDO.
Approximately 60% of brain kynurenine arises from the blood (Reference Gal and Sherman68) where it is further metabolised to either the excitotoxic metabolite quinolinic acid, which acts as an agonist at NMDA glutamate receptors, or to the neuroprotective metabolite kynurenic acid that acts as an antagonist at the NMDA receptor (Reference Myint and Kim44). Under non-inflammatory/non-stress conditions there is a balance between the end products of the neurodegenerative and neuroprotective pathways (Reference Perkins and Stone69).
The distribution of the tryptophan–kynurenine pathway in the brain is concentrated mainly in the astrocytes and microglia (Reference Grant and Kapoor70). In the astrocytes, kynurenine is mainly synthesised to kynurenic acid by kynurenine aminotransferase whereas in the microglia it is primarily converted to 3-hydroxykynurenine and quinolinic acid (Reference Guillemin, Smythe, Takikawa and Brew71,Reference Guillemin, Smith and Kerr72). Under non-inflammatory conditions the end product of kynurenine metabolism is kynurenic acid but following the activation of the microglia by stress or inflammation the neurodegenerative pathway predominates (Reference Guillemin, Wang and Brew73). This is further enhanced under inflammatory conditions by the apoptosis of the astrocytes by quinolinic acid (Reference Guillemin, Wang and Brew73). This results in the exposure of neurons to the exotoxic kynurenine metabolites thereby contributing to the decrease in the brain volume reported to occur in patients with chronic schizophrenia or depression (Reference Bremner, Narayan, Anderson, Staib, Miller and Charney74,Reference van Erp, Saleh and Huttunen75). The regions of the brain involved in the metabolism of kynurenine may differ according to the pathophysiology of the psychiatric disorder. In major depression Steiner et al. (Reference Steiner, Walter and Gos76) have shown that the quinolinic acid concentration in the microglia from subregions of the anterior cingulated gyrus is increased. This evidence that the excitotoxic pathway is increased in depression is further supported by the increase in plasma 3-hydroxykynurenine (Reference Myint, Schwarz and Muller77).
Thus as a consequence of immune activation the changes in the tryptophan–kynurenine pathway play a major role in the dysfunctional neurotransmitter systems in the brain and, in addition, contribute to the changes in the brain structure and function which characterise depression (Fig. 3).

Fig. 3 The tryptophan–kynurenine pathway that links depression, pain and inflammation. IDO, indoleamine 2,3-dioxygenase in spleen, blood, kidney and brain; TDO, tryptophan 2,3-dioxygenase in liver; IFN, interferons; IL4, interleukin-4; ATP, adenosine-tri-phosphate; NAD, nicotinamide adenine dinucleotide; NMDA, N-methyl-d-aspartate: 1=kynureninase; 2=kynurenine oxygenase; 3=kynurenine aminotransferase.
In recent years, attention has centred on the neurotoxic consequences of the increase in quinolinic acid and the intermediates formed from kynurenine in the tryptophan–kynurenine pathway. While such neurotoxins undoubtedly play a critical role in the neurodegenerative changes associated with chronic psychiatric disorders such as depression and schizophrenia, it is often overlooked that quinolinic acid is also an important substrate for the formation of nicotinamide adenine dinucleotide (NAD+). As NAD+ is a key component of the respiratory chain, chronic pathological changes that reduce its formation are liable to have adverse consequences for intermediary metabolism particularly in neurons that are critically dependent on high energy sources. It is estimated that ~99% of tryptophan that is not used for protein and serotonin synthesis is metabolised to NAD+ via the tryptophan–kynurenine pathway, and therefore this pathway is important for the synthesis of this vital cofactor (Reference Gal and Sherman68,Reference Han, Cai, Tagle and Li78). This situation would be compounded by a reduction in the availability of insulin, a key factor in the transport of glucose into neurons (Reference Oxenkrug79). As there is evidence that insulin receptor resistance is a frequent feature of depression, and other major psychiatric disorders and with age-related pathology associated with the dementias (Reference Lee, Tong, Hang, Deochand and de la Monte80), it seems reasonable to conclude that a chronic decrease in high energy substrates resulting from a deficit in glucose and essential cofactors may be of crucial importance in understanding the causes of increased neuronal apoptosis (Reference Lee, Tong, Hang, Deochand and de la Monte80).
This situation is further complicated by mitochondrial dysfunction in depression which results in a decrease in the synthesis of ATP and related high energy molecules, combined with an increase in oxidative damage. In addition, the synthesis of superoxide radicals results from a decrease in the ratory chain and causes increases in the damage to the mitochondrial membranes by opening the permeability transition pores (Reference Sas, Robotka, Toldi and Vecsei81). The oxygen free radical synthesis is further enhanced by xanthenuric acid and 3-hydroxykynurenine which are formed in the brain as a result of the inflammation enhanced tryptophan–kynurenine pathway.
Possible mechanisms of action of antidepressants as anti-inflammatory agents
The monoamine hypothesis of depression, that has formed the basis for an understanding of the psychopathology of depression for the past 50 years, has concentrated attention on dysfunction of the serotonergic, noradrenergic and dopaminergic systems (Reference Maes and Meltzer82–Reference Wong, Kling and Munson85). More recently, attention has been directed to the role of neuropeptides, such as corticotropin-releasing factor and BDNF (Reference Duman, Heninger and Nestler38,Reference Manji, Drevets and Charney86). However, an important shift in emphasis has occurred in the past 2 decades with the discovery that a dysfunctional immune and endocrine system play an increasingly important role in major psychiatric disorders.
If the pathological basis of depression is linked to a dysfunctional immune, endocrine and neurotransmitter system, how does this hypothesis link with the monaminergic theory of depression and the mode of action of antidepressants? It is widely acknowledged that antidepressants increase the intracellular concentration of cyclic adenosine monophosphate (cAMP) by activating the G-protein coupled monoamine receptors (Reference Malberg and Blendy87) and also by reducing the activity of intraneuronal phosphodiesterase which catalyses cAMP (Reference Wong, Whelan and Deloukas88). It is also evident from ex vivo and in vitro studies that antidepressants have immunosuppressive effects (Reference Castanon, Leonard, Neveu and Yirmiya89–Reference Xia, DePierre and Nassberger91), an effect which has also been explained as a consequence of an increase in intracellular cAMP (Reference Xia, DePierre and Nassberger91). This explanation seems credible in the light of the demonstration that inhibitors of phosphokinase A and cAMP synthesis block the effects of antidepressants on the release of IL and NO from activated microglia (Reference Hashioka, Klegeris and Monji92). It must be emphasised that these results were obtained from in vitro studies in which the IC 50 values for the antidepressants were high (in the range of 26–51 μM) in comparison to the estimated in vivo concentration for the therapeutic effects. However, Baumann et al. (Reference Baumann, Hiemke and Ulrich93) have reported that psychotropic drugs accumulate in the brain in concentration that are 10–40 times higher than in the serum which must be taken into account when extrapolating changes seen in vitro to those occurring under therapeutic conditions.
As Anisman et al. (Reference Anisman, Merali and Hayley94) emphasised in their substantial review, that it is unlikely that the multiple neurochemical changes associated with depression act independently of one another and more likely that they reflect a cascade of neuronal changes, some of which reflect reactions to chronic stress, an important trigger for depression. A consequence of physical or psychological stress in those predisposed to depression is the activation of the inflammatory pathways. Thus if the pathological changes in depression are attributable to an impairment of neuronal structure and function, it can be hypothesised that anti-inflammatory drugs, and drugs that inhibit the synthesis of NO and reactive oxygen species, would exhibit antidepressant activity. There is experimental (Reference Harkin, Bruce, Craft and Paul95–Reference Wegener and Volke98) and clinical evidence (Reference Muller99) to support the evidence that cyclooxygenase (COX) and nitric oxide synthase (NOS) inhibitors have antidepressant-like activity. Experimentally minocycline, an antibiotic with antioxidant properties, that inhibits activated microglia (Reference Ekdahl, Claasen, Bonde, Kokaia and Lindvall100) has also been shown to restore hippocampal neurogenesis, an effect which correlates in a reduction in depressive-like behaviour.
NOS inhibitors have also been shown to exhibit antidepressant properties in rodent models of depression (Reference Harkin, Bruce, Craft and Paul95,Reference Wegener and Mathé97,Reference Wegener and Volke98,Reference Joca and Guimaraes101–Reference Harkin, Connor, Walsh, St John and Kelly104). Such studies give support to the hypothesis that drugs that reduce the neurotoxic challenge caused by proinflammatory cytokines, NO, prostaglandin E2 and other neurotoxins have potential antidepressant action.
The precise mechanism whereby antidepressants reduce the release of cytokines from activated macrophages and microglia is uncertain. However, there is experimental evidence that the selective serotonin re-uptake inhibitor (SSRI) antidepressants, in vitro and in relatively high concentrations (5 μM), inhibit the release of TNF-α and NO from activated microglia (Reference Horikawa, Kato and Mizoguchi105). The activation of microglia by IFN-γ is associated with an influx of calcium ions that triggers the intracellular changes (phosphokinase C, p38, MAPK, ERK 1 and 2 and the JAK-STAT pathway) which result in the release of the proinflammatory cytokines. Thus by impeding the influx of calcium ions, the SSRI’s reduce the activation of the JAK-STAT pathway and thereby reduce the inflammatory challenge. These effects are unrelated to the action of the SSRIs on the serotonin transporter.
Hashioka et al. (Reference Hashioka, Klegeris and Monji92) have shown that the activation of murine microglia in vitro by IFN-γ, which led to an increase in IL-6 and NO, was attenuated by imipramine, fluvoxamine and reboxetine. Lithium had a paradoxical effect, however in that it inhibited the synthesis of NO but increased the synthesis of the proinflammatory cytokines! However, this would not appear to be a common effect of all antidepressants. Thus Horikawa et al. (Reference Horikawa, Kato and Mizoguchi105) demonstrated that neither buproprion nor agomelatine, that are effective antidepressants which lack a SSRI antidepressant profile, inhibit the release of cytokines from activated microglia. Furthermore O’Sullivan et al. (Reference O’Sullivan, Ryan, Curtin, Harkin and Connor106) also failed to demonstrated that atomoxetine and desipramine inhibited the release of TNF-γ and NO. Conversely, the atypical antipsychotic aripiprazole, which is used as an adjunctive treatment in therapy resistant depression (Reference Weber, Lyseng-Williamson and Scott107), did inhibit the release of TNF-α from activated microglia (Reference Kato, Mizoguchi and Monji108). Thus it would appear that the inhibition of activated microglia may be important in the mode of action of some types of antidepressants but would not appear to be a unifying mechanism of action of all antidepressants.
Prostaglandins as a cause of inflammation in depression; are anti-inflammatory drugs therefore potential antidepressants?
The prostaglandins are a group of lipophilic molecules that are synthesised from membrane bound arachidonic acid by the sequential actions of cyclo-oxygenases 1 and 2, and the respective prostaglandin synthases. They produce their physiological effects by activating G-protein coupled receptors, prostaglandin D receptor and EP 1–4 subtypes of the prostaglandin E receptors. Studies of prostaglandin receptor knockout mice have demonstrated that the prostenoids play a major role in the transition to, and maintenance of, chronic inflammation (Reference Honda, Segi-Nishida, Miyachi and Narumiya109,Reference Narumiya110).
Of the two major cyclo-oxygenases involved in the formation of prostaglandins, COX 2 is induced by lipopolysaccharides in the brain and by proinflammatory cytokines such as IL1 and β, IL-6, cytokines that are known to be increased in depressed patients. Unlike COX 2, COX 1 is constitutively expressed independently of these inflammatory stimuli. In addition, there is now evidence that the prostaglandins, once formed, are able to prolong acute inflammatory responses by initiating chronic gene dependent expression by facilitating proinflammatory cytokines and suppressing the differentiation of the anti-inflammatory producing Th2 cells. The prostaglandins also contribute to the inflammatory cascade by inducing the synthesis of inflammatory chemokines and in facilitating the infiltration of inflammatory cells into inflammatory sites (Reference Aoki and Narumiya111). Whether such a process is involved in psychiatric disorders in which inflammation is not site specific is unknown but it has been established in the case of focussed inflammatory lesions such as intracranial aneurysms (Reference van Gijn, Kerr and Rinkel112).
What is the relative importance of the COX enzymes in initiating and maintaining inflammation in depression? COX 1 is constitutively expressed in most tissues including the brain and is considered to be the enzyme primarily responsible for the homoeostatic synthesis of prostaglandins (Reference Phillis, Horrocks and Farooqui113) whereas COX 2 is mainly induced in response to inflammatory stimuli. This has led to the development of selective COX 2 inhibitors that are considered to reduce inflammation without reducing the physiologically important COX 1-derived prostaglandins. However, in the brain COX 2 is expressed in the hippocampus and in glutamatergic neurons where it plays a role in maintaining synaptic function, long-term synaptic plasticity and neurovascular coupling during functional hyperaemia (Reference Niwa, Araki, Morham, Ross and Iadecola114).
While the development of drugs that suppress the synthesis of PGE2 has long been recognised, it is often overlooked that PGE2 has an important physiological role in tissue maintenance and regeneration. Recent research has demonstrated that, in mice, inhibition of 15-hydroxyprostaglandin dehydrogenase (15-PGDH), which metabolises PGE2, increases tissue regeneration (Reference Zhang, Desai and Yang115). These researchers report that deleting 15-PGDH, results in haematopoeisis, liver regeneration, resistance to inflammation and reduction in the ulceration of the gastrointestinal tract. As there are concerns that COX 2 inhibitors can cause adverse cardiovascular effects, and may exacerbate inflammation of the gastrointestinal tract in Crohn’s disease, it seems timely to consider the development of drugs that are specific inhibitors of 15-PGDH as anti-inflammatory agents.
In experimental studies it has been shown that COX 2 is linked to anti-inflammatory and neuroprotective changes (Reference Bertolini, Ottani and Sandrini116). This raises possible concern regarding the long-term effects of COX 2 inhibitors, such as celecoxib, particularly as COX 1 is largely unaffected by COX 2 inhibitors and continue to contribute to the inflammatory changes (Reference Pepicelli, Fedele and Berardi117). There is also evidence that the inhibition of COX 2 increases the gene expression of inflammatory factors such as microsomal PGE2 synthase-1, TLR 2, monocyte chemotactic protein-1 (MCP-1) in vascular cells in the brain and also in parenchymal microglia following the systemic administration of LPS (Reference Blais, Turrin and Rivest118). In addition, the integrity of the BBB is reduced by inflammation and whereas some COX 2 inhibitors are ineffective in counteracting these effects, selective COX 1 inhibitors have been shown to reduce the disruptive effect of proinflammatory cytokines, such as TNF-α, on the integrity of the BBB.
These effects of the COX inhibitors are mainly based on in vitro and acute experimental studies, and therefore their relevance to clinical studies in which the COX 2 inhibitors have been demonstrated to facilitate the therapeutic effects of antidepressants (Reference Muller119) must be treated with caution. Nevertheless such observations do stress the importance of evaluating the potential adverse effects of COX inhibitors should they be used long-term in the treatment of depression.
If, as appears from experimental and clinical studies, that COX 2 inhibitors have a limited effect on the chronic inflammatory response, how can the antidepressant-like potential of drugs such as celecoxib be explained? The lack of correlation between the CSF-PGE2 concentration and the reduction in the severity of the clinical symptoms following the administration of COX 2 inhibitors to patients with multiple sclerosis (Reference Mattsson, Yaong and Rosengren120) suggest that their therapeutic effects may reside in the enhancement of anti-inflammatory mediators rather than in the reduction of PGE2. In support of this view, it is known that membrane bound archidonic acid, besides being an important substrate for PGE2, is also metabolised to anti-inflammatory products such as epoxyeicostrienoic acid and lipoxins (Reference Ahmad, Rose and Vagni121). This may help to explain the clinical observation that the COX 2 inhibitor celecoxib, and aspirin, augment the antidepressant response of reboxetine (Reference Muller119) and other antidepressants (Reference Mendlewicz, Kriwin, Oswald, Souery, Alboni and Brunello122).
Thus COX 2 inhibitors may afford neuroprotection without primarily reducing the PGE2 induced inflammatory effects. Indeed, novel COX 2 derived eicosanoids such as lipoxin A4, the E and D series of the resolvins, and the protectins with anti-inflammatory actions have been shown to be the end products of COX 2, but not COX 1, pathway (Reference Serhan and Chiang123), and can metabolise the endogenous endocannabinoids anadamide and 2-arachidonylglycerol (Reference Kozak, Crews and Morrow124) which modify immune function and enhance neurogenesis (Reference Wolf and Ullrich125). Thus it can be surmised that any neuroprotective effect of COX 2 inhibitors could be attributable to their indirect effect on anadamide and non-prostaglandin metabolites rather than to a selective attenuation of PGE2 synthesis. COX 2 is generally regarded as an enzyme responsible for the synthesis of the important inflammatory mediator, PGE2, which contributes to tissue injury!
However, there is considerable evidence that chronic inhibition of COX 2 may have beneficial actions involving the prostanoid signalling pathway. The prostaglandin E2 receptor (EP2) has been implicated in some of the physiological and pathological changes attributed to COX 2, the nature of these changes depending on whether the G-protein independent, or G-protein dependent, pathway is activated by PGE2. As Jiang and Dingledine (Reference Jiang and Dingledine126) have indicated in their recent review, the activation of EP2 receptors in neurons promote cAMP-protein kinase A dependent neuroprotection whereas, conversely, EP2 activation in the microglia, via the cyclic AMP/Epac pathway, mediates the activation of proinflammatory mediators, such as inducible NOS, COX 2, NADPH oxidase and many proinflammatory cytokines. This results in tissue injury.
Thus the EP2 receptor can exert both beneficial or deleterious effects in the brain which may reflect the complexity of the therapeutic outcome when COX 2 inhibitors are used. The neural action of EP2 receptor agonists promote acute neuroprotection, neuronal survival and neuronal plasticity whereas activation of the EP2 receptors in microglia result in secondary neurotoxicity due to the activation of COX 2 and inducible NOS. This results in chronic brain inflammation, neuronal apoptosis, oxidative stress, etc. Thus the PGE2-EP2 pathway plays a multiple role in inflammation, tumorogenesis, cytoprotection or neurodegeneration. Perhaps future anti-inflammatory antidepressant targets involving the neuronal EP2 receptors require consideration!
Despite the experimental evidence that the action of COX 2 inhibitors is more complicated than was envisaged in terms of the inhibition of the synthesis of PGE2, there is also experimental evidence that COX 2 inhibitors can selectively modulate monoamine metabolism in the rat cortex. Johansson et al. (Reference Johansson, Falk, Marcus and Svensson127) demonstrated that celecoxib, when administered concurrently with either reboxetine, a selective noradrenaline re-uptake inhibitor, or the SSRI antidepressant fluoxetine, enhanced the release of noradrenaline and dopamine, and serotonin, respectively. This could be another explanation for the synergistic effect of COX 2 inhibitors on the therapeutic effects of antidepressants.
Besides celecoxib, other types of anti-inflammatory drugs have also been shown to have antidepressant-like effects associated with their anti-inflammatory actions For example, minocycline is an antibiotic which reduces the release of proinflammatory cytokines from activated microglia. Minocycline has anti-inflammatory profile and experimental evidence of antidepressant like effects (Reference Molina-Hernandez, Tellez-Alcantara, Perez-Garcia, Olivera-Lopez and Jaramillo-Jaimes128,Reference Levine, Cholestoy and Zimmerman129). Similarly, the anti-inflammatory and TNF-α antagonist infliximab, used in the treatment of psoriasis, Crohn’s disease and related chronic inflammatory conditions, has mood-elevating effects that occur before the beneficial change in the pathological symptoms of these inflammatory disorders.
Thus even if the further development of anti-inflammatory drugs based on the inhibition of the cyclo-oxygenases is precluded due to their relatively limited therapeutic efficacy and potential adverse side effects, there is sufficient experimental and clinical evidence to suggest that the modulation of other pathways involved in neuroinflammation could provide important therapeutic targets in the future.
Towards an integrated view of the mechanism of action of antidepressants
From the evidence presented here, it is apparent that there are a multitude of changes in which underlie the pathology of depression. The recent emphasis on the broader metabolic changes that are coincidental with the core symptoms of depression has contributed to a wider understanding of the importance of the immune and endocrine axes in the pathology of the condition. This has stimulated research into the role of proinflammatory cytokines as important triggers for depression and illustrates how they may integrate the endocrine and neurotransmitter changes with the impact of inflammation on the neuron. Further, this approach helps to explain the chronic outcome of depression, particularly in elderly patients, where dementia frequently occurs as a consequence of the neurodegenerative cascade initiated by the proinflammatory cytokines, a reduction in neurotrophic factors (e.g. BDNF) due to hypercortisolaemia and an increase in reactive oxygen and nitrogen species. In addition there is now evidence that the end products of the inflammation activated tryptophan–kynurenine pathway (such as 3-hydroxykynurenine and quinolinic acid) also play a substantial role in the neurodegenerative changes seen in chronic major depression. These aspects have been discussed in detail elsewhere (Reference Myint and Kim44,Reference Leonard and Myint62).
Conclusion
Despite the limitations, there is sufficient evidence to conclude that the chronic, low grade, inflammation hypothesis contributes towards a comprehensive explanation of the psychopathology of depression and how antidepressants may alleviate the symptoms. More importantly, the hypothesis identifies possibilities for the development of new types of antidepressants which, hopefully, will also improve their efficacy.
Acknowledgements
The author express his thanks to Ayemu Myint, Cai Song and the colleagues who have contributed to my studies in psychoimmunology mentioned in this review.
Conflicts of Interest
None.



