
Contents
Research Article
Studio della Coagulazione normale del Sangue nei Gemelli con il Metodo tromboelastografico
-
- Published online by Cambridge University Press:
- 01 August 2014, pp. 1-19
-
- Article
-
- You have access
- Export citation
-
In the course of a research work based on the study of the thrombelastogram in 20 male and 20 female MZ pairs, and in 20 male and 20 female DZ pairs, the Authors have evaluated the genetic impact on the thrombelastographic traits. The analysis of the experimental data shows that the following data are especially genetically influenced:
1) the maximum width (¾)
2) the elasticity modulus (
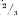 )
)3) the time r (½)
4) the thrombelastographic index (½)
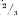 )
)Hereditary Affections of the Retina and Choroid
-
- Published online by Cambridge University Press:
- 01 August 2014, pp. 20-68
-
- Article
-
- You have access
- Export citation
Agénésie Sacrococcygienne et Syndrome de Bonnevie-Ullrich Étude Génétique et Chromosomique
-
- Published online by Cambridge University Press:
- 01 August 2014, pp. 69-89
-
- Article
-
- You have access
- Export citation
The Occurrences of symmetrical and asymmetrical Terminations of Main Line D, C, B and A in Mohammedans of Rajasthan
-
- Published online by Cambridge University Press:
- 01 August 2014, pp. 90-98
-
- Article
-
- You have access
- Export citation
Correction
Errata Corrige
-
- Published online by Cambridge University Press:
- 01 August 2014, p. 98
-
- Article
-
- You have access
- Export citation
In memoriam
Tage Kemp 28/VIII/1896 – 7/1/1964
-
- Published online by Cambridge University Press:
- 01 August 2014, pp. 99-100
-
- Article
-
- You have access
- Export citation
Recensioni
A. Franceschetti, J. François, J. Babel: Les hérédo-dégénérescences chorio-rétiniennes (Dégénérescences tapéto-rétiniennes), Rapport présenté à la Societé Française d'Ophtalmologie le 7 mai 1963, avec de nombreuses figures en noir et 53 planches hors-texte en couleurs, Tome Premier, Masson & C.ie Editeurs, Paris 1963.
-
- Published online by Cambridge University Press:
- 01 August 2014, p. 101
-
- Article
-
- You have access
- Export citation
Rigomar Rieger, Die Genommutationen (Ploidiemutationen), 183 Seiten, 72 Abbildungen, Preis DM 23,90. Verlag Gustav Fischer, Jena 1963.
-
- Published online by Cambridge University Press:
- 01 August 2014, pp. 101-102
-
- Article
-
- You have access
- Export citation
F. Vacirca, La teoria generale della patologia. Ediz. Minerva Medica, Torino 1962. 1° Volume di pagg. xix+626, L. 10.000.
-
- Published online by Cambridge University Press:
- 01 August 2014, pp. 102-103
-
- Article
-
- You have access
- Export citation
Ilse Schwidetzky: Das Menschenbild der Biologie. Ergebnisse und Probleme der naturwissenschaftlichen Anthropologie. Gustav Fischer Verlag - Stuttgart, 1959. VIII+218 pp., 81 illustrazioni. DM 24.
-
- Published online by Cambridge University Press:
- 01 August 2014, pp. 103-104
-
- Article
-
- You have access
- Export citation
E. Chieppa: Paramorfismi (Scoliosi-Cifosi-Lordosi) e tecnica del trattamento. Tipografia & Libreria « Unione Biellese ».
-
- Published online by Cambridge University Press:
- 01 August 2014, p. 104
-
- Article
-
- You have access
- Export citation
Books Received
Libri Ricevuti
-
- Published online by Cambridge University Press:
- 01 August 2014, p. 104
-
- Article
-
- You have access
- Export citation
Front matter
AMG volume 13 issue 1 Cover and Front matter
-
- Published online by Cambridge University Press:
- 01 August 2014, pp. f1-f6
-
- Article
-
- You have access
- Export citation