


INTRODUCTION
The animal trypanosomiases (or trypanosomoses) include a variety of wasting diseases caused by unicellular protozoan parasites of the genus Trypanosoma (order Kinetoplastida). All relevant animal pathogenic trypanosomes (T. vivax – subgenus Duttonella, T. congolense – subgenus Nannomonas and T. brucei spp. – subgenus Trypanozoon) (Fig. 1) belong to the Salivaria group (Haag et al. Reference Haag, O'hUigin and Overath1998), so-called because their transmission to the vertebrate host occurs principally via the infected saliva of blood-sucking insects. Most valuable domestic livestock (bovines, ovines, caprines, equids, camelids and suids) are susceptible to infection with one or more of these Trypanosoma species. This can lead to acute and/or chronic forms of wasting disease, causing high morbidity, mortality and infertility in the absence of treatment (Leach and Roberts, Reference Leach and Roberts1981; Connor, Reference Connor1992). By affecting agricultural production and animal husbandry, the animal trypanosomiases have a high economic and social impact in vast areas of the tropics and subtropics where transmission occurs. Africa has historically suffered the greatest burden (Steverding, Reference Steverding2008), but the negative effects are also increasing in South America and South-East Asia, where unrestricted animal movements favour the spread of some trypanosome species.

Fig. 1. Morphological characteristics of the bloodstream form trypomastigote of the three most important livestock trypanosomes. T. brucei group trypanosomes (T. b. brucei, T. b. evansi, T. b. equiperdum) are morphologically indistinguishable (with the exception of the non-proliferative stumpy-form in T. b. brucei). The trypomastigote is the disease-relevant form and the target of therapy.
Chemotherapy and chemoprophylaxis represent the mainstay of animal trypanosomiases control, ensuring animal health and production in enzootic countries. However, the available veterinary trypanocides (Table 1) are inadequate and outmoded. Only six compounds are currently licensed, and their narrow therapeutic indices restrict their use, especially when even low-level resistance arises. By far, the most usage is of two compounds, diminazene aceturate and isometamidium chloride, largely applied against animal trypanosomiases in Africa (Holmes et al. Reference Holmes, Eisler, Geerts, Maudlin, Holmes and Miles2004), with suramin also being relatively widely used to treat T. b. evansi infections. Worryingly, an increasing number of reports of resistance to this handful of existing chemicals, particularly diminazene and isometamidium, indicate their future utility to be in jeopardy (Geerts et al. Reference Geerts, Holmes, Eisler and Diall2001; Delespaux and de Koning, Reference Delespaux and de Koning2007).
Table 1. Currently available veterinary trypanocides.
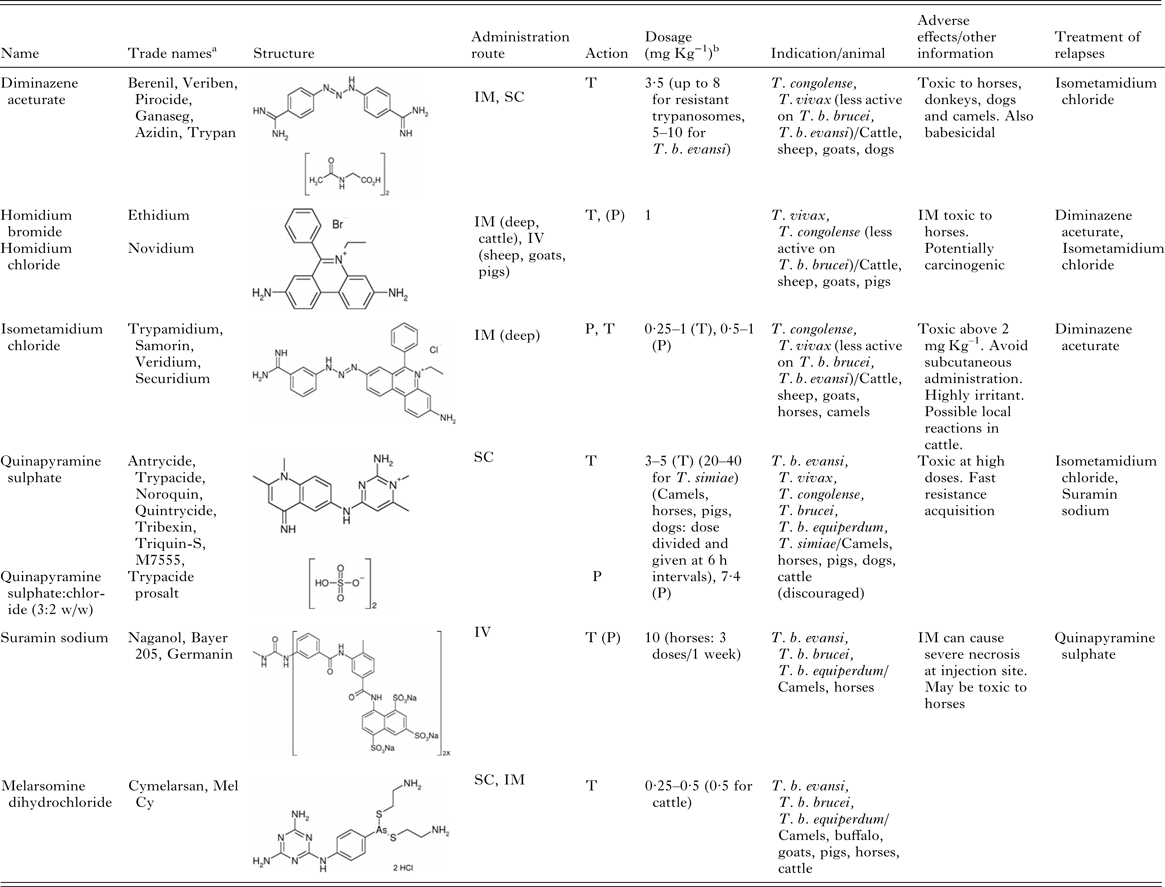
IM, intramuscular; IV, intravenous; SC, subcutaneous; T, therapeutic action; P, prophylactic action. Note. Products used in animals producing meat or milk for human consumption should only be used in full compliance with product labels including withdrawal periods.
a The list of the trade names is not complete.
b Dosages are for single administration except were stated otherwise.
It has been estimated that as many as 35 million doses of trypanocides are used annually in sub-Saharan Africa alone (Holmes, Reference Holmes2013), which represents a figure suitable to treat only around one-third of the cattle at risk (Swallow, Reference Swallow2000). Inclusion of trypanocides sold informally in the African market may substantially increase the total number of doses sold annually, which may be as high as 70 million doses (Frans van Gool, personal communication, 2015). Despite this demand, the high costs of drug development and the low anticipated profit from the sale of chemotherapeutics in developing countries have disincentivized commercial pharmaceutical investments in this field (Connor, Reference Connor1992). In recent years, a public–private partnership, GALVmed (Global Alliance for Livestock Veterinary Medicines), supported by funding from the Bill & Melinda Gates Foundation and the UK Department for International Development, has emerged to fill the gap, and has committed to the development of new therapeutic and prophylactic trypanocidal drugs (http://www.galvmed.org/en/). However, even in the best case scenario, a novel licensed compound is unlikely to be available for several years yet; hence the rational, correct use of the trypanocides already available is of paramount importance.
THE ANIMAL TRYPANOSOMIASES: DISTRIBUTION, TRANSMISSION, HOSTS, PATHOLOGY AND ECONOMIC IMPACT
Animal African trypanosomiasis (AAT, nagana)
AAT [also called nagana, from the Zulu word ‘N'gana’ which means ‘powerless/useless’ (Steverding, Reference Steverding2008)], is caused by trypanosome species T. congolense, T. vivax and, to a lesser extent, T. brucei spp. (Fig. 1). The disease is widespread in sub-Saharan Africa (Fig. 2), where it is cyclically transmitted by the tsetse fly (Glossina spp.), the same vector responsible for the transmission of human-infective trypanosomes (T. brucei gambiense and T. b. rhodesiense, the aetiological agents of human African trypanosomiasis, HAT, or sleeping sickness) (Barrett et al. Reference Barrett, Burchmore, Stich, Lazzari, Frasch, Cazzulo and Krishna2003). In animals, tsetse flies can also transmit trypanosomes mechanically when they begin a blood meal on an infected host and end it on another one, provided that the time between the two meals is short enough to ensure survival of parasites in the insect mouthparts, as shown in experimental infections in goats (Moloo et al. Reference Moloo, Kabata and Gitire2000). Unlike other trypanosomes, T. vivax does not multiply in the tsetse midgut, but remains confined to the insect proboscis, where it completes its short life cycle (Gardiner, Reference Gardiner1989). This is the reason why this species can also be transmitted mechanically by other haematophagous flies, in particular horseflies (Tabanus spp.) and stable flies (Stomoxys spp.). Mechanical transmission has allowed T. vivax to spread far beyond the limits of the African tsetse belt: this parasite is now established in Mauritius and in 13 South American countries (Fig. 2), where it probably arrived in the 18th or 19th century via infected Zebu cattle exported from West Africa (Jones and Davila, Reference Jones and Davila2001; Osorio et al. Reference Osorio, Madruga, Desquesnes, Soares, Ribeiro and Costa2008), an origin corroborated by phylogenetic studies (Cortez et al. Reference Cortez, Ventura, Rodrigues, Batista, Paiva, Anez, Machado, Gibson and Teixeira2006). Although T. vivax remains enzootic in South America primarily due to mechanical transmission, other potential modes of transmission include perinatal and iatrogenic routes or via alternative, as yet unidentified vectors (Osorio et al. Reference Osorio, Madruga, Desquesnes, Soares, Ribeiro and Costa2008). This lack of definitive knowledge greatly hampers the implementation of surveillance and control strategies (Jones and Davila, Reference Jones and Davila2001). Non-tsetse transmitted T. vivax infection in cattle is also recognized in parts of Africa, for example in regions of Ethiopia, Chad and Sudan (Ahmed et al. Reference Ahmed, Rahman, Hassan, Salih, Paone and Cecchi2016). Mechanical transmission of T. congolense has been shown under experimental conditions (Desquesnes and Dia, Reference Desquesnes and Dia2003) and can therefore not be excluded from contributing to its spread in Africa (Desquesnes et al. Reference Desquesnes, Biteau-Coroller, Bouyer, Dia and Foil2009).
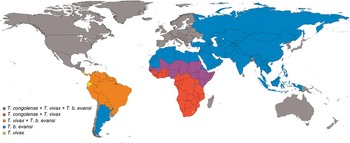
Fig. 2. Countries where the most important livestock trypanosomes are present. Modified from (Auty et al. Reference Auty, Torr, Michoel, Jayaraman and Morrison2015), based on PubMed search and including countries where data were not available and parasite presence is inferred. To note that the real geographical distribution in some countries is limited (as, for example, for T. congolense in South Africa, Namibia and Botswana and for T. b. evansi in Russia). Cases of eradicated outbreaks of T. b. evansi in Europe (i.e. in France) are not indicated.
The host range is wide (Uilenberg, Reference Uilenberg1998). Trypanosoma congolense is considered the most pathogenic trypanosome in cattle (followed by T. vivax), but it also causes infections in horses, sheep, goats, pigs and dogs. Apart from bovines, T. vivax can affect sheep, goats, horses and camels (Osorio et al. Reference Osorio, Madruga, Desquesnes, Soares, Ribeiro and Costa2008). Trypanosoma b. brucei is found in various domestic ungulates but it is particularly virulent in dogs, camels and horses, the latter often succumbing to infection within a few months in the absence of treatment. In areas where more than one trypanosome species is present, mixed infections in domestic animals are often encountered (Kihurani et al. Reference Kihurani, Nantulya, Mbiuki, Mogoa, Nguhiu-Mwangi and Mbithi1994; Auty et al. Reference Auty, Mundy, Fyumagwa, Picozzi, Welburn and Hoare2008; Biryomumaisho et al. Reference Biryomumaisho, Rwakishaya, Melville, Cailleau and Lubega2013; Takeet et al. Reference Takeet, Fagbemi, De Donato, Yakubu, Rodulfo, Peters, Wheto and Imumorin2013; Moti et al. Reference Moti, De Deken, Thys, Van Den Abbeele, Duchateau and Delespaux2015) and modern molecular techniques (Desquesnes and Davila, Reference Desquesnes and Davila2002) facilitate speciation. Many wild animal species in Africa also host one or more trypanosome species and can serve as reservoirs for both human and domestic animal infective trypanosomes (Mulla and Rickman, Reference Mulla and Rickman1988; Auty et al. Reference Auty, Anderson, Picozzi, Lembo, Mubanga, Hoare, Fyumagwa, Mable, Hamill, Cleaveland and Welburn2012). Similarly, wild South American fauna can harbour T. vivax and act as reservoir of infection (Osorio et al. Reference Osorio, Madruga, Desquesnes, Soares, Ribeiro and Costa2008).
Belonging to the same Nannomonas subgenus as T. congolense, T. simiae is the only trypanosome species to be extremely pathogenic to pigs, which represent the main host, although other domestic species can harbour the parasite (Joshua and Kayit, Reference Joshua and Kayit1984; Salim et al. Reference Salim, Bakheit and Sugimoto2014). In pigs, T. simiae causes a hyperacute, often fatal infection, with death often occurring within 48 h of the appearance of symptoms (Leach and Roberts, Reference Leach and Roberts1981). For this reason, chemoprophylaxis is preferred to curative treatment.
The pathogenicity of trypanosomal infections varies considerably depending on several factors, including parasite-related aspects (species and virulence), host (species, breed, age, immunological status, nutritional status, presence of co-infection and physical condition), vector (species, density, infection rate and host preference), epidemiological situation (endemic or epidemic) and the environment (e.g. the availability of food and water and the season) (Leach and Roberts, Reference Leach and Roberts1981; Van den Bossche and Delespaux, Reference Van den Bossche and Delespaux2011). Anaemia is the most prominent pathological feature of AAT (Taylor and Authié, Reference Taylor, Authié, Maudlin, Holmes and Miles2004) and, in conjunction with other systemic lesions, can contribute to death through eventual congestive heart failure. Other symptoms include pyrexia, lymph node and spleen enlargement, ataxia, lethargy, weight loss, oedema, immunosuppression, abortion and decrease in milk production. The immunosuppression caused by trypanosomes can affect animal health by interfering with vaccination against other diseases (Singla et al. Reference Singla, Juyal and Sharma2010), or by increasing susceptibility of the host to other infections. Inflammatory, degenerative lesions are also observed, and can damage various organs such as heart, central nervous system (CNS), eyes, testes, ovary and pituitary gland. Death may occur within weeks from onset of the acute disease. Otherwise the animal enters a chronic phase (spontaneous recovery is rare but not unknown), characterized by intermittent or sub-patent parasitaemia, general malaise and infertility, and may last months or years prior to death (Taylor and Authié, Reference Taylor, Authié, Maudlin, Holmes and Miles2004).
While mortality due to the disease is clearly important, the impact upon overall cultivation and crop production due to reduced draught power is the most significant contributor to the economic impact of AAT (Swallow, Reference Swallow2000). This is considered the livestock disease with the highest impact on agricultural production and animal husbandry in Africa, causing annual losses which run to billions of US$ (Shaw et al. Reference Shaw, Cecchi, Wint, Mattioli and Robinson2014). Across the tsetse belt as many as 55 million cattle are at risk of infection (Cecchi and Mattioli, Reference Cecchi and Mattioli2009), plus 30 million sheep and 40 million goats. Of these cattle, 3 million die every year from AAT. The disease has devastating effects on the livelihoods of local farmers, for whom cattle represent not only a source of food (meat and milk), manure, and draught power, but have also fundamental social roles as ‘living banks’ and are used for social obligations (e.g. dowry and ritual use) (Swallow, Reference Swallow2000; Grace et al. Reference Grace, Randolph, Affognon, Dramane, Diall and Clausen2009; Mungube et al. Reference Mungube, Vitouley, Allegye-Cudjoe, Diall, Boucoum, Diarra, Sanogo, Randolph, Bauer, Zessin and Clausen2012).
Infection with T. vivax is considered an emerging disease in South America where it has a significant impact on cattle farming, but where it also affects horses and other ruminants (Batista et al. Reference Batista, Riet-Correa, Teixeira, Madruga, Simoes and Maia2007, Reference Batista, Oliveira, Rodrigues, Damasceno, Oliveira, Alves, Paiva, Brito, Medeiros, Rodrigues and Teixeira2009, Reference Batista, Rodrigues, Olinda, Silva, Vale, Camara, Reboucas, Bezerra, Garcia and Teixeira2012; Da Silva et al. Reference Da Silva, Garcia Perez, Costa, Franca, De Gasperi, Zanette, Amado, Lopes, Teixeira and Monteiro2011). In a region including the Brazilian Pantanal and the Bolivian lowlands, where cattle ranching is the single most important economic activity (11 million head of cattle are reared in the region), the losses caused to the industry by a single outbreak of T. vivax in 1995 were calculated at more than US$ 160 million (Seidl et al. Reference Seidl, Davila and Silva1999). The gross financial burden of T. vivax in South America, however, is not known with any degree of certainty.
Surra
Surra (from the Hindi word for ‘rotten’) is the most widely used of a plethora of names given to T. b. evansi infection in animals (Desquesnes et al. Reference Desquesnes, Holzmuller, Lai, Dargantes, Lun and Jittaplapong2013b ). As seen for T. vivax, T. b. evansi (a T. brucei subspecies) has also evolved a mechanical mechanism of transmission that has allowed this species to spread beyond Africa by export of infected animals (Lun et al. Reference Lun, Lai, Li, Lukes and Ayala2010). Trypanosoma b. evansi is today the pathogenic animal trypanosome with the broadest geographical distribution (Fig. 2), which stretches from North-East Africa to much of Asia in the east (Luckins, Reference Luckins1988; Payne et al. Reference Payne, Sukanto, Djauhari, Partoutomo, Wilson, Jones, Boid and Luckins1991; Lun et al. Reference Lun, Fang, Wang and Brun1993) and to Latin America in the west (Desquesnes et al. Reference Desquesnes, Holzmuller, Lai, Dargantes, Lun and Jittaplapong2013b ), and it is spreading steadily. In Europe, recent imported cases of surra have been documented and vigilance remains necessary after outbreaks in the Canary Islands, mainland Spain, France and Germany (Desquesnes et al. Reference Desquesnes, Bossard, Patrel, Herder, Patout, Lepetitcolin, Thevenon, Berthier, Pavlovic, Brugidou, Jacquiet, Schelcher, Faye, Touratier and Cuny2008; Gutierrez et al. Reference Gutierrez, Desquesnes, Touratier and Buscher2010; Tamarit et al. Reference Tamarit, Tejedor-Junco, Gonzalez, Alberola and Gutierrez2011; Defontis et al. Reference Defontis, Richartz, Engelmann, Bauer, Schwierk, Buscher and Moritz2012).
Several probable or suggested methods of surra transmission exist: by biting insects including horseflies and stable flies (the major credited route), by vampire bats, by iatrogenic (e.g. as a result of a vaccination intervention), sexual, horizontal or vertical transmission, or by per-oral contamination in the case of carnivores eating infected meat (Desquesnes et al. Reference Desquesnes, Dargantes, Lai, Lun, Holzmuller and Jittapalapong2013a ).
Trypanosoma b. evansi can parasitize a wide range of wild and domestic animal hosts, but the infection is particularly pathogenic in horses, camels and Asian water buffaloes (Desquesnes et al. Reference Desquesnes, Holzmuller, Lai, Dargantes, Lun and Jittaplapong2013b ). There is increasing evidence that common rodents are an important reservoir host for T. b. evansi and other trypanosomes (Jittapalapong et al. Reference Jittapalapong, Inpankaew, Sarataphan, Herbreteau, Hugot, Morand and Stich2008; Maia da Silva et al. Reference Maia da Silva, Marcili, Ortiz, Epiphanio, Campaner, Catao-Dias, Shaw, Camargo and Teixeira2010; Kocher et al. Reference Kocher, Desquesnes, Yangtara, Morand and Jittapalapong2015; Pumhom et al. Reference Pumhom, Morand, Tran, Jittapalapong and Desquesnes2015), such as T. lewisi, a parasite of rats also found in atypical human infections (Howie et al. Reference Howie, Guy, Fleming, Bailey, Noyes, Faye, Pepin, Greenwood, Whittle, Molyneux and Corrah2006; Sarataphan et al. Reference Sarataphan, Vongpakorn, Nuansrichay, Autarkool, Keowkarnkah, Rodtian, Stich and Jittapalapong2007). These findings revive the important question of rodents as reservoirs of other T. brucei species. Rare cases of human infection with T. b. evansi (Joshi et al. Reference Joshi, Shegokar, Powar, Herder, Katti, Salkar, Dani, Bhargava, Jannin and Truc2005; Haridy et al. Reference Haridy, El-Metwally, Khalil and Morsy2011; Van Vinh et al. Reference Van Vinh, Buu, Desquesnes, Herder, Phu Huong, Campbell, Van Cuong, Yimming, Chalermwong, Jittapalapong, Ramon Franco, Tri Tue, Rabaa, Carrique-Mas, Pham Thi Thanh, Tran Vu Thieu, Berto, Thi Hoa, Van Minh Hoang, Canh Tu, Khac Chuyen, Wills, Tinh Hien, Thwaites, Yacoub and Baker2016), where individuals were infected through trypanosome-carrying animal blood, have been reported and, in at least one case, infection was associated with a null mutation in the trypanosome lytic factor blood component Apolipoprotein L1 (APOL1), which normally protects humans from animal trypanosome infections (Vanhollebeke et al. Reference Vanhollebeke, Truc, Poelvoorde, Pays, Joshi, Katti, Jannin and Pays2006; Truc et al. Reference Truc, Buscher, Cuny, Gonzatti, Jannin, Joshi, Juyal, Lun, Mattioli, Pays, Simarro, Teixeira, Touratier, Vincendeau and Desquesnes2013). In a more recent case, no mutations in APOL1 were found to explain the unusual infection (Van Vinh et al. Reference Van Vinh, Buu, Desquesnes, Herder, Phu Huong, Campbell, Van Cuong, Yimming, Chalermwong, Jittapalapong, Ramon Franco, Tri Tue, Rabaa, Carrique-Mas, Pham Thi Thanh, Tran Vu Thieu, Berto, Thi Hoa, Van Minh Hoang, Canh Tu, Khac Chuyen, Wills, Tinh Hien, Thwaites, Yacoub and Baker2016).
Symptoms of surra overlap those previously described for AAT and their intensity can vary greatly between and within host species and depend on the geographical area and epidemiological situation (Desquesnes et al. Reference Desquesnes, Holzmuller, Lai, Dargantes, Lun and Jittaplapong2013b ).
In the Philippines, outbreaks of surra cause high morbidity and mortality in water buffaloes and other large ruminants, greatly affecting the livelihood of local small-scale farmers (Dargantes et al. Reference Dargantes, Mercado, Dobson and Reid2009; Desquesnes et al. Reference Desquesnes, Dargantes, Lai, Lun, Holzmuller and Jittapalapong2013a ). In the Brazilian Pantanal T. b. evansi affects over 6000 horses per year (of the 50 000 present), with serious consequences to the local economy, horses being essential for herding livestock. The total impact of T. b. evansi infection in horses in this region was estimated at US$ 2·4 million per year (Seidl et al. Reference Seidl, Moraes, Aguilar and Silva1998). Surra is also one of the most frequent diseases affecting camels in North Africa, causing severe economic damage.
Dourine
Dourine is a disease caused by the subspecies T. brucei equiperdum, the only Salivarian trypanosome whose transmission cycle avoids invertebrate vectors completely. Instead, this parasite is transmitted among horses and other equids during mating (Claes et al. Reference Claes, Buscher, Touratier and Goddeeris2005). Of note, vertical or perinatal transmission of trypanosomes other than T. b. equiperdum in the reproductive tissues has been reported (Griffin, Reference Griffin1983; Melendez et al. Reference Melendez, Forlano and Figueroa1993; Lindner and Priotto, Reference Lindner and Priotto2010; Biteau et al. Reference Biteau, Asencio, Izotte, Rousseau, Fevre, Pillay and Baltz2016), although the role and relative importance of this mode of transmission in the field is not clear.
Trypanosoma b. equiperdum is an important veterinary trypanosome endemic in Africa and Asia, and is also found in the Middle-East, South-East Europe and South America. Strict control policies have eradicated T. b. equiperdum from Western Europe in the past century (Claes et al. Reference Claes, Buscher, Touratier and Goddeeris2005), but the risk of reintroduction remains, as shown by a recent outbreak in Italy (Pascucci et al. Reference Pascucci, Di Provvido, Camma, Di Francesco, Calistri, Tittarelli, Ferri, Scacchia and Caporale2013).
The infection presents with typical oedema of the genital organs as well as weakness, emaciation, urethral discharge, characteristic plaques in the skin and neurological symptoms such as lack of coordination of the hind legs (Hagos et al. Reference Hagos, Goddeeris, Yilkal, Alemu, Fikru, Yacob, Feseha and Claes2010). Dourine in horses is generally fatal without treatment but it is usually subclinical in donkeys and mules (Brun et al. Reference Brun, Hecker and Lun1998).
Considering the transmission mechanism and the absence of a reservoir in other species, the control strategies for the disease follow a different approach as compared with other insect-borne forms of trypanosomiasis (Claes et al. Reference Claes, Buscher, Touratier and Goddeeris2005). The World Health Organization for Animal Health (OIE) recommends breeding and movement restrictions, compulsory notification and slaughter of infected animals to block new infection outbreaks or achieve eradication. Additionally, pharmacological therapy is not advised as this may result in clinical improvement but not in complete cure, leaving the animal as a potential carrier of the parasite. However, the feasibility or effectiveness of this strict policy in developing countries, where horses have a significant role in transport and agriculture, is questionable. Here, chemotherapy may help to sustain animal health and productivity. Although no official cure for dourine is available, studies have indicated the efficacy of melarsomine in the treatment of acute and chronic T. b. equiperdum infection in horses (Hagos et al. Reference Hagos, Goddeeris, Yilkal, Alemu, Fikru, Yacob, Feseha and Claes2010).
ANIMAL TRYPANOSOME SPECIES: VIRULENCE, TISSUE DISTIBUTION, BIOLOGY AND LABORATORY TOOLS
Trypanosoma congolense and T. simiae
Trypanosoma congolense is the smallest of the pathogenic trypanosomes (see Fig. 1 for its morphology). The species is divided into three main subgroups (i.e. Savannah, Forest and Kilifi) based on molecular markers (Hide and Tait, Reference Hide, Tait, Maudlin, Holmes and Miles2004; Auty et al. Reference Auty, Torr, Michoel, Jayaraman and Morrison2015), the Savannah subgroup being the most virulent (Bengaly et al. Reference Bengaly, Sidibe, Boly, Sawadogo and Desquesnes2002a , Reference Bengaly, Sidibe, Ganaba, Desquesnes, Boly and Sawadogo b ) and the most clinically important in cattle. However, even within the same Savannah subgroup substantial differences in virulence exist, with some strains causing only mild infections (Masumu et al. Reference Masumu, Marcotty, Geysen, Geerts, Vercruysse, Dorny and Van den Bossche2006), highlighting the complexity and subtlety of the balance between the level of parasite persistence and the host immune system.
In the vertebrate host, T. congolense parasites remain confined to the vascular system, where they bind to circulating erythrocytes (Banks, Reference Banks1979) and to endothelial cells (Hemphill et al. Reference Hemphill, Frame and Ross1994) through their flagellum, causing damage at the adhesion site (Banks, Reference Banks1980). Attachment of the bloodstream form is also observed in in vitro culture, where parasites adhere to the bottom of the flask, a phenotype unique to T. congolense among trypanosome species (Coustou et al. Reference Coustou, Guegan, Plazolles and Baltz2010).
Today, long-term culture of the pathogenic bloodstream form is possible only for a limited number of strains (e.g. IL3000 and STIB910) (Coustou et al. Reference Coustou, Guegan, Plazolles and Baltz2010). Genetic tools have been developed for this species, including a gene overexpression system (Coustou et al. Reference Coustou, Guegan, Plazolles and Baltz2010) and RNA interference (although, in this case, only for the procyclic insect form) (Inoue et al. Reference Inoue, Otsu, Ferraro and Donelson2002; Coustou et al. Reference Coustou, Guegan, Plazolles and Baltz2010). A draft genome sequence of strain IL3000 has also been published (Jackson et al. Reference Jackson, Berry, Aslett, Allison, Burton, Vavrova-Anderson, Brown, Browne, Corton, Hauser, Gamble, Gilderthorp, Marcello, McQuillan, Otto, Quail, Sanders, van Tonder, Ginger, Field, Barry, Hertz-Fowler and Berriman2012) and offers the potential to accelerate discovery of biomarkers for diagnosis and targets for new drugs. However, despite the veterinary importance of T. congolense, the data available to understand its biology and pathogenicity and, therefore, to improve treatment, are scanty. It appears that this parasite has a carbohydrate metabolism that differs significantly from that of the far more widely studied T. brucei (Agosin and von Brand, Reference Agosin and von Brand1954), with indications of a more pronounced mitochondrial activity in its bloodstream form. These dissimilarities may have relevance in the very different responses of these species to trypanocides (Leach and Roberts, Reference Leach and Roberts1981) and in the identification of potential drug targets. Of note, T. congolense lacks an orthologue of the T. brucei TbAT1 gene that encodes the P2 nucleoside transporter (see subsection Diminazene aceturate below), which is central to the uptake of the trypanocidal drug diminazene (Munday et al. Reference Munday, Rojas Lopez, Eze, Delespaux, Van Den Abbeele, Rowan, Barrett, Morrison and de Koning2013). Trypanosoma congolense has a correspondingly reduced sensitivity to diminazene, which is not accumulated to the same degree in these parasites.
Similarly, the closely related T. simiae does not easily infect common laboratory rodents and, therefore, little data on this organism is available. However, a method for the axenic in vitro culture of the bloodstream form of this parasite has been published (Zweygarth et al. Reference Zweygarth, Moloo, Kaminsky and Gray1992), offering the means to accelerate our ability to dissect the parasite's biology.
Trypanosoma vivax
Among African trypanosomes, T. vivax (Fig. 1) is the most phylogenetically distinct species (Fig. 3). Specific isolates present with different pathogenicity in cattle, in some cases causing chronic, sub-clinical infections and in others acute, haemorrhagic infections (Wellde et al. Reference Wellde, Chumo, Adoyo, Kovatch, Mwongela and Opiyo1983; Magona et al. Reference Magona, Walubengo and Odimin2008).
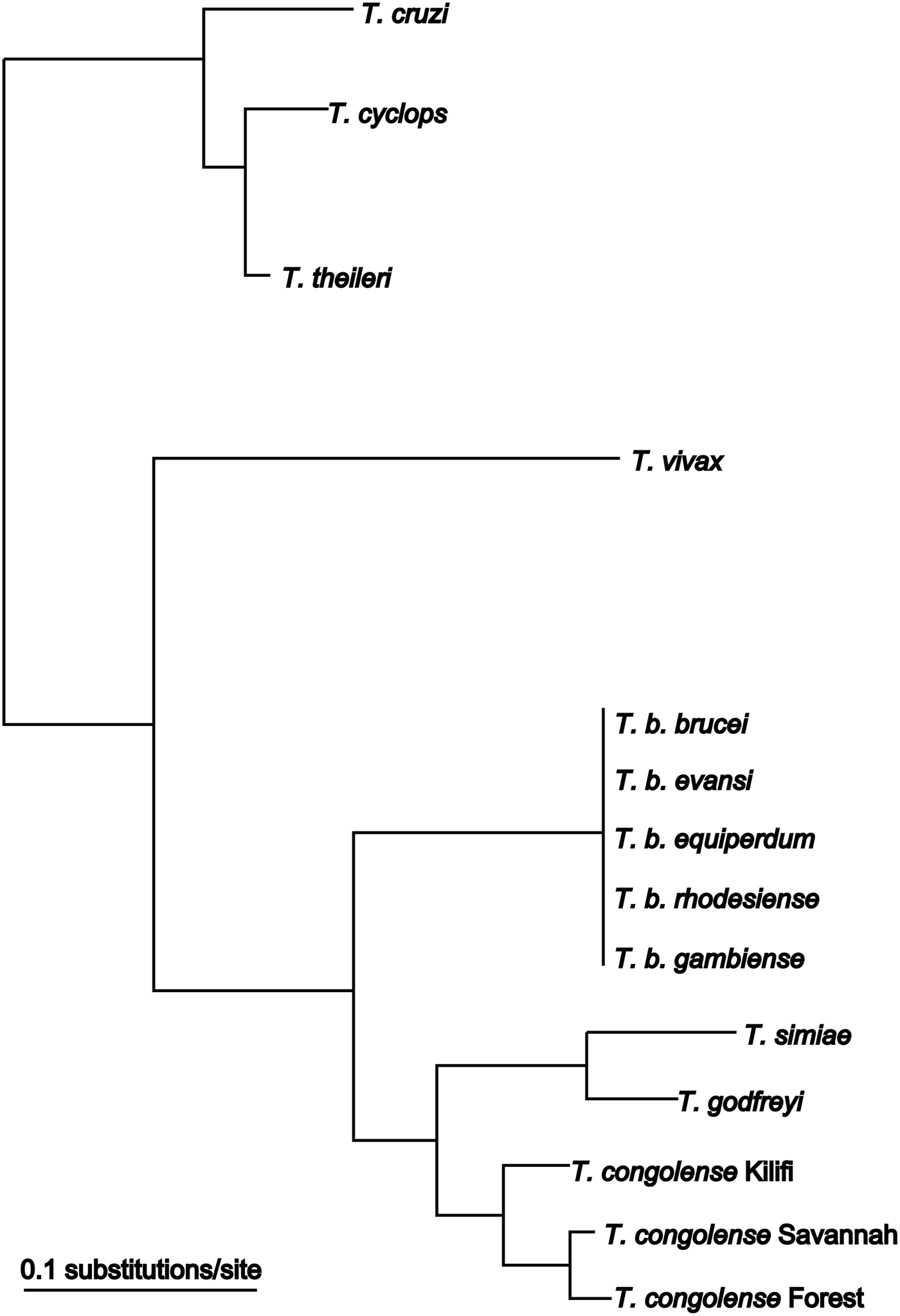
Fig. 3. Phylogenetic tree based on SSU rRNA sequences from trypanosome species. Modified from (Cortez et al. Reference Cortez, Ventura, Rodrigues, Batista, Paiva, Anez, Machado, Gibson and Teixeira2006).
Although T. vivax (as T. congolense) has been considered typically to remain confined to the vascular system of the host, some strains may, especially in late infections, also reach extravascular locations (e.g. lymph nodes, eyes and cerebrospinal fluid) where they may directly damage tissues and where they are less accessible to drug treatment (Whitelaw et al. Reference Whitelaw, Gardiner and Murray1988; Osorio et al. Reference Osorio, Madruga, Desquesnes, Soares, Ribeiro and Costa2008; D'Archivio et al. Reference D'Archivio, Cosson, Medina, Lang, Minoprio and Goyard2013).
Trypanosoma vivax is generally difficult to cultivate in the laboratory and this has restricted biological studies into this parasite. Short-term, axenic culture systems for the bloodstream form have been reported (Brun and Moloo, Reference Brun and Moloo1982; Zweygarth et al. Reference Zweygarth, Gray and Kaminsky1991; D'Archivio et al. Reference D'Archivio, Medina, Cosson, Chamond, Rotureau, Minoprio and Goyard2011) but they have been difficult to reproduce in other laboratories and have not entered routine use. Most studies on this trypanosome species are, therefore, conducted in in vivo laboratory models; however, very few T. vivax strains have been isolated that readily infect rodents and most published in vivo work on this species comprises the very few mouse-infective strains, the main one being Y486 and its derivatives (Gibson, Reference Gibson2012). A simplified system for in vitro cultivation of the insect form of T. vivax was recently described and genetic manipulation methodology implemented (D'Archivio et al. Reference D'Archivio, Medina, Cosson, Chamond, Rotureau, Minoprio and Goyard2011). As with T. congolense, studies into the biochemical physiology of T. vivax have lagged behind those in T. brucei but significant differences with the metabolism of bloodstream form T. brucei were clear from early studies (Desowitz, Reference Desowitz1956), which probably explains incongruence in potency of different chemical classes against these species.
Trypanosoma brucei spp.
Trypanosoma brucei spp. (Fig. 1) include both animal (T. b. brucei, T. b. evansi, T. b. equiperdum) and human (T. b. rhodesiense, T. b. gambiense) infective subspecies. Unlike T. vivax (most strains at least) or T. congolense, T. brucei group trypanosomes are found in both the vascular system and in other tissues, and can parasitize the brain in experimental infections (Moulton, Reference Moulton1986; Grab and Kennedy, Reference Grab and Kennedy2008; Coles et al. Reference Coles, Myburgh, Ritchie, Hamilton, Rodgers, Mottram, Barrett and Brewer2015); descriptions of this clinical condition in field settings are limited, other than for equids, which are particularly susceptible to T. brucei (Tuntasuvan et al. Reference Tuntasuvan, Sarataphan and Nishikawa1997; Ranjithkumar et al. Reference Ranjithkumar, Saravanan, Yadav, Kumar, Singh and Dey2014). As the most widely used drugs to treat animal trypanosomes (diminazene and isometamidium) do not cross the blood–brain barrier, the presence of parasites in sites other than the bloodstream represents a potentially important issue for treatment of T. brucei. Parasites from inaccessible body sites including the CNS may eventually re-establish infection in the bloodstream and cause relapse following treatment with these drugs (Myburgh et al. Reference Myburgh, Coles, Ritchie, Kennedy, McLatchie, Rodgers, Taylor, Barrett, Brewer and Mottram2013). Trypanosoma b. equiperdum is quite unique, it being mainly a tissue parasite, found in the capillaries of the urogenital tract and rarely in peripheral blood (Brun et al. Reference Brun, Hecker and Lun1998). This makes diagnosis, parasite isolation and treatment particularly difficult.
Trypanosoma b. brucei is the most extensively studied trypanosome. Some lineages (e.g. Lister 427) are well adapted to laboratory in vitro culture and have been used as model organism to study many eukaryotic cell processes. The genome of this species was published in 2005 (Berriman et al. Reference Berriman, Ghedin, Hertz-Fowler, Blandin, Renauld, Bartholomeu, Lennard, Caler, Hamlin, Haas, Bohme, Hannick, Aslett, Shallom, Marcello, Hou, Wickstead, Alsmark, Arrowsmith, Atkin, Barron, Bringaud, Brooks, Carrington, Cherevach, Chillingworth, Churcher, Clark, Corton and Cronin2005) and its metabolism has been widely studied (Shameer et al. Reference Shameer, Logan-Klumpler, Vinson, Cottret, Merlet, Achcar, Boshart, Berriman, Breitling, Bringaud, Butikofer, Cattanach, Bannerman-Chukualim, Creek, Crouch, de Koning, Denise, Ebikeme, Fairlamb, Ferguson, Ginger, Hertz-Fowler, Kerkhoven, Maser, Michels, Nayak, Nes, Nolan, Olsen and Silva-Franco2015). It has long been known that, in its bloodstream form, T. brucei species depend entirely on glycolysis for energy production, while Krebs cycle and oxidative phosphorylation are active only in the insect stages. New, comprehensive metabolomics approaches (Creek et al. Reference Creek, Mazet, Achcar, Anderson, Kim, Kamour, Morand, Millerioux, Biran, Kerkhoven, Chokkathukalam, Weidt, Burgess, Breitling, Watson, Bringaud and Barrett2015) are modifying this paradigm and, in conjunction with transcriptomic approaches, a clearer understanding of trypanosome metabolism is emerging.
Trypanosoma b. evansi and T. b. equiperdum can be considered petite mutants of T. brucei, so named after petite mutants of yeast that have lost mitochondrial respiratory function. These parasites have lost part (dyskinetoplastic parasites) or all (akinetoplastic parasites) of their kinetoplast DNA (kDNA), which constitutes the mitochondrial genome and comprises a network of circular concatenated mini- and maxi-circles (Schnaufer et al. Reference Schnaufer, Domingo and Stuart2002; Lai et al. Reference Lai, Hashimi, Lun, Ayala and Lukes2008). Although long considered as two separate species, it has been proposed that T. b. evansi and T. b. equiperdum be reclassified as subspecies of T. brucei, based on phylogenetic analysis of sequenced genomes (Carnes et al. Reference Carnes, Anupama, Balmer, Jackson, Lewis, Brown, Cestari, Desquesnes, Gendrin, Hertz-Fowler, Imamura, Ivens, Koreny, Lai, MacLeod, McDermott, Merritt, Monnerat, Moon, Myler, Phan, Ramasamy, Sivam, Lun, Lukes, Stuart and Schnaufer2015), and we have adopted this convention here. As the kinetoplast genome encodes for an essential subunit (F0-A6) of the mitochondrial F1F0 ATP synthase, T. b. evansi and T. b. equiperdum cannot complete their life cycle in the fly and are locked in the trypomastigote stage, which relies on glycolysis for ATP production. A compensating mutation in the nuclear genome-encoded γ-subunit of the ATP synthase allows these parasites to maintain their mitochondrial membrane potential irrespective of the F0-A6 subunit and, therefore, to survive in the absence of the kinetoplast genome (Dean et al. Reference Dean, Gould, Dewar and Schnaufer2013). It is for this reason that these parasites lost their dependency on the tsetse fly for transmission.
CONTROL STRATEGIES AND TRYPANOTOLERANCE
All of the important livestock trypanosomes described above are extracellular parasites in mammals and evade the host immune defences by continuously changing their surface coat (Horn, Reference Horn2014), one of the immune-evading mechanisms that essentially preclude the development of conventional vaccines (La Greca and Magez, Reference La Greca and Magez2011; Cnops et al. Reference Cnops, Magez and De Trez2015). Hence, control of animal trypanosomiases relies primarily on the use of insecticides or traps to control the vector (especially in the case of tsetse-transmitted trypanosomiases), and on the use of trypanocides to control the parasite (Holmes, Reference Holmes2013). (The control strategy for dourine follows a completely different approach and has been described separately; see subsection Dourine above). Since vector control can be expensive when used on a large scale and is not always sustainable or effective, administration of trypanocidal drugs represents the main intervention tool in most poor rural endemic areas, ensuring maximum effects at relatively little cost (Grace et al. Reference Grace, Randolph, Affognon, Dramane, Diall and Clausen2009; Van den Bossche and Delespaux, Reference Van den Bossche and Delespaux2011). The cost-effectiveness of this practice was shown both in Africa (at least under certain circumstances) (Shaw et al. Reference Shaw, Wint, Cecchi, Torr, Mattioli and Robinson2015) and elsewhere (Seidl et al. Reference Seidl, Moraes, Aguilar and Silva1998, Reference Seidl, Davila and Silva1999; Dobson et al. Reference Dobson, Dargantes, Mercado and Reid2009). Control of parasites with chemotherapeutic and chemoprophylactic agents has the double effect of limiting the losses caused by the infection and of eliminating the transmissible trypanosome reservoir (Welburn et al. Reference Welburn, Beange, Ducrotoy and Okello2015). Effective treatment of the acute phase of infection usually leads to prompt recovery of the animal; the use of trypanocides in the chronic phase, however, usually clears parasitaemia, but clinical recovery in these instances may require a significantly longer time, depending on the severity of symptoms such as weight loss and organ damage.
Some indigenous African livestock breeds (e.g. N'Dama, Muturu and Dahomey) are more resistant to trypanosome infection than imported breeds (classically temperate ‘European’ taurine breeds but also including Asian-derived Bos indicus breeds, relatively new to trypanosome endemic areas, such as Boran). This phenomenon is called ‘trypanotolerance’ and is defined as the ‘capacity to survive and remain productive after trypanosome infection’ (Murray et al. Reference Murray, Morrison and Whitelaw1982). A major factor enabling these animals to cope with trypanosome infections is a better capacity to limit both anaemia and parasitaemia (Naessens, Reference Naessens2006). The use of trypanotolerant breeds has helped livestock productivity in various endemic regions in Africa and elsewhere, and it is often advocated as an important control strategy. Wild animals, which have co-evolved with trypanosomes, are also usually trypanotolerant and rarely suffer from clinical disease when infected.
TREATMENT STRATEGIES AND CHALLENGES
Treatment and prophylaxis of pathogenic trypanosome infections in animals relies on only six compounds (Table 1), most dating back to the first half of the 20th century (Leach and Roberts, Reference Leach and Roberts1981). Moreover, several factors limit their use. The current drugs all have small therapeutic indices and can also cause local irritancy at the injection site. Most importantly, extensive utilization in the past has led to the appearance of resistant parasites in the field, and the fact that many of these trypanocides are chemically related has exacerbated the situation with cross-resistance onset (Peregrine, Reference Peregrine1994). A number of currently used compounds appear to target the kinetoplast, causing its loss (Shapiro and Englund, Reference Shapiro and Englund1990; Chitambo and Arakawa, Reference Chitambo and Arakawa1992b ), but the actual mode of action of these trypanocides and the biochemical mechanisms underpinning resistance are largely unclear. As noted above, differences in biochemical physiology and host organ distribution discriminate each of the veterinary trypanosomes and, therefore, the different trypanocides have divergent ability to kill based on specific potency against each species and pharmacokinetic parameters affecting distribution.
Most trypanocides have therapeutic rather than prophylactic activity, but the phenanthridine isometamidium is mostly used for its prophylactic effects (Stevenson et al. Reference Stevenson, Sones, Gicheru and Mwangi1995). Unfortunately, these drugs are less active against T. b. evansi (Toro et al. Reference Toro, Leon, Lopez, Pallota, Garcia and Ruiz1983) and are less used outside of sub-Saharan Africa (Reid, Reference Reid2002). The decision as to whether to use therapeutic or prophylactic drugs depends on several factors, including the risk of infection, drug availability and distribution logistics (Gu et al. Reference Gu, Gettinby, McKendrick, Murry, Peregrine and Revie1999). Ideally, in areas of low prevalence, only those animals that present with clinical disease attributable to trypanosomes and/or have confirmed infection should be treated with therapeutic drugs; instead, in areas of high challenge, prophylactic drugs applied to the whole herd are more cost-effective, providing much greater reduction of mortality and morbidity and avoiding the adverse effects of infection on productivity (Gu et al. Reference Gu, Gettinby, McKendrick, Murry, Peregrine and Revie1999). Single-dose therapeutic and prophylactic products for cattle are preferred, as multiple-dose administration regimens are often not practical in developing countries, where animal handling facilities are typically very limited.
As new compounds are not likely to become available in the near future (i.e. the most optimistic outlook is at least 3–5 years before a new compound could realistically be expected to be registered through current initiatives), prudent use of those already on the market is paramount. However, in field settings drug usage is often difficult to monitor and regulate. In hyperendemic African countries, trypanocides are usually administered directly by farmers, who can easily obtain them at local markets for a relatively affordable price (for less than US$ 1 per treatment). Unfortunately, most livestock keepers in the affected regions have limited access to tools which (a) enable accurate diagnosis, and frequently farmers are reliant solely on clinical signs, which are often not pathognomonic; and (b) provide information or training regarding optimal drug usage and dosage, and this combination of factors can lead to drug misuse (Van den Bossche et al. Reference Van den Bossche, Doran and Connor2000; Grace et al. Reference Grace, Randolph, Affognon, Dramane, Diall and Clausen2009). Moreover, in an unregulated market, poor quality or counterfeit trypanocides are widespread in some areas, especially in Africa, where documented product specifications are scarce (Sutcliffe et al. Reference Sutcliffe, Skellern, Araya, Cannavan, Sasanya, Dungu, van Gool, Munstermann and Mattioli2014; Tchamdja et al. Reference Tchamdja, Kulo, Akoda, Teko-Agbo, Assoumy, Niang, Batawui, Adomefa, Bankole, Kombiagou, Hoppenheit, Clausen, Mattioli, Peter, Napier, De Deken, Marcotty, Van Den Abbeele and Delespaux2016). To improve veterinary drug standards and tackle the issue of counterfeit drugs two laboratories for trypanocide quality control checks were recently set up in Africa (one in Dakar and one in Dar Es Salaam) thanks to a GALVmed-FAO (Food and Agriculture Organization of the United Nations) initiative with other collaborating partners (Sutcliffe et al. Reference Sutcliffe, Skellern, Araya, Cannavan, Sasanya, Dungu, van Gool, Munstermann and Mattioli2014).
Besides correct dosage administration, various other options to extend the life of current trypanocides exist. Different approaches (such as delivery systems including complexing to polymeric substances promoting slow release or alternative formulations) have been considered in order to improve therapeutic efficacy (Peregrine, Reference Peregrine1994; Geerts et al. Reference Geerts, Brandt and De Deken1999; Kroubi et al. Reference Kroubi, Karembe and Betbeder2011; Unciti-Broceta et al. Reference Unciti-Broceta, Arias, Maceira, Soriano, Ortiz-Gonzalez, Hernandez-Quero, Munoz-Torres, de Koning, Magez and Garcia-Salcedo2015). These could allow the use of lower quantities of trypanocide in a more effective way and, consequently, pose a decreased risk of toxicity and possibly decreased resistance development.
Unlike the situation with HAT, where the nifurtimox–eflornithine combination therapy (NECT) is now the preferred first line treatment for second-stage disease (Priotto et al. Reference Priotto, Kasparian, Mutombo, Ngouama, Ghorashian, Arnold, Ghabri, Baudin, Buard, Kazadi-Kyanza, Ilunga, Mutangala, Pohlig, Schmid, Karunakara, Torreele and Kande2009; Alirol et al. Reference Alirol, Schrumpf, Amici Heradi, Riedel, de Patoul, Quere and Chappuis2013), no drug combinations are currently used for the animal trypanosomiases. Instead, alternating use of compounds, particularly diminazene and isometamidium (called a ‘sanative pair’), with low risk of cross-resistance, is recommended where possible. In particular, in the case of relapse the animal should be treated with a different drug class from the one previously administered, in order not to reinforce drug resistance selection (Leach and Roberts, Reference Leach and Roberts1981). Due to the chemical relatedness of several veterinary trypanocides, however, this approach is not always practicable. Thus, in order to maintain the efficacy of the currently used compounds, it is important that chemotherapeutic and chemoprophylactic dosage regimens are rationalized on the basis of the drug-susceptibility phenotype of trypanosome populations in a given locality. However, such rationalization is not possible, because the systems that are currently available to characterize the drug resistance phenotype of trypanosome populations are not field applicable (Peregrine, Reference Peregrine1994). Limited numbers of field isolates can be characterized and all of the systems take many months to provide definitive data (see section Tests for resistance detection below). There is therefore a requirement for new assays that will rapidly quantify the drug resistance phenotype of large numbers of trypanosome isolates.
VETERINARY TRYPANOCIDES: DOSAGE, PHARMACOKINETICS, MODE OF ACTION AND RESISTANCE
Diminazene aceturate
Diminazene aceturate (Table 1) was introduced for the treatment of babesiosis and African trypanosomiasis in livestock in 1955. It belongs to the diamidine class of compounds, a member of which (pentamidine) has also been used for HAT since the 1930s (Steverding, Reference Steverding2010). Ironically, it was pursuing a structure-activity iterative synthesis from a compound belonging to a different class, Surfen C [at the time of its introduction in the 1930s, the best available agent against T. congolense infections (Bennett, Reference Bennett1936)], that led to diminazene development (Hawking, Reference Hawking, Schnitzer and Hawking1963). Although it was anti-T. congolense activity in experimental rodents that initially drove development, today's in vitro systems, where anti-parasite potency can be tested without confounding issues related to pharmacokinetic behaviour in hosts, show that diminazene is substantially less potent against T. congolense than it is against T. brucei group trypanosomes. This feature is attributable to the fact that its uptake into the latter parasites via the P2/TbAT1 transporter (see later) allows concentrative and rapid uptake (De Koning et al. Reference De Koning, Anderson, Stewart, Burchmore, Wallace and Barrett2004). In T. congolense, which lacks an orthologue of TbAT1 (Munday et al. Reference Munday, Rojas Lopez, Eze, Delespaux, Van Den Abbeele, Rowan, Barrett, Morrison and de Koning2013), uptake is less robust, explaining its lower activity.
Diminazene is today the most commonly used trypanocide in cattle, sheep and goats, due to its activity against both T. congolense and T. vivax and its relatively low toxic side effects. The compound also effectively cures surra and is, for example, the mainstay of treatment of T. b. evansi in the Philippines (Reid, Reference Reid2002). The recommended therapeutic dose is 3·5 mg kg−1 body weight for AAT due to T. congolense and T. vivax (7 mg kg−1 may be recommended against resistant isolates) and 7 mg kg−1 is indicated for AAT due to T. brucei and for surra, administered by intramuscular or subcutaneous injection (Connor, Reference Connor1992). The common practice of administering 3·5 mg kg−1 of the drug to treat T. b. evansi infections is considered an underdosing, and this misuse may have contributed to the emergence of resistant strains in South-East Asia (Desquesnes et al. Reference Desquesnes, Dargantes, Lai, Lun, Holzmuller and Jittapalapong2013a ). The fact that higher doses appear to be needed to treat T. brucei group trypanosomes, in spite of these parasites being more sensitive to the drug, probably relates to their wider tissue dispersal compared with T. congolense and T. vivax, underlining the key role of host pharmacokinetics.
Diminazene is only applied as a curative agent and is not used for prophylaxis, as it is rapidly metabolized and excreted (Peregrine and Mamman, Reference Peregrine and Mamman1993). After rapid absorption (the peak blood level is reached within 1 h of dosing), elimination follows a biphasic or triphasic behaviour depending on the animal species and formulation; elimination half-life values following intramuscular administration varied from 11–19 h in sheep and goats, to 74 to >200 h in cattle (Mamman et al. Reference Mamman, Aliu and Peregrine1993; Peregrine and Mamman, Reference Peregrine and Mamman1993; Mdachi et al. Reference Mdachi, Murilla, Omukuba and Cagnolati1995; El Banna et al. Reference El Banna, Abo el-Sooud and Soliman1999). Cattle excrete diminazene mainly in the urine, together with two main metabolites: p-aminobenzamidine and p-amino-benzamide (Kellner et al. Reference Kellner, Eckert and Volz1985). Diminazene residues may persist for several weeks in the edible tissues of cattle and other food-producing animals, especially in the liver and kidney, whereas the drug levels in milk peak at 6 h and fall to below detection limits after 48 h (FAO, 1990). For this reason it is advised that cattle and sheep destined for human consumption are subject to a 21–35 days pre-slaughter withdrawal (discard) from drug, while a 3-day milk discard period is recommended (FAO, 1990; Peregrine and Mamman, Reference Peregrine, Gray and Moloo1993); however, product-specific withdrawal periods as given on product labels should be adhered to.
The trypanocidal mode of action of diminazene has not been completely elucidated. The compound binds the minor groove of the DNA at AT-rich sites (Wilson et al. Reference Wilson, Tanious, Mathis, Tevis, Hall and Boykin2008). In trypanosomes, the kDNA is a known target of the drug, and kDNA binding can cause inhibition of replication and kDNA loss (Shapiro and Englund, Reference Shapiro and Englund1990), possibly exacerbated by an inhibitory effect on mitochondrial type II topoisomerase (Portugal, Reference Portugal1994). It had long been believed that loss of the kinetoplast might not be sufficient to kill trypanosomes, as viable dyskinetoplastic strains do occur naturally and also can be produced artificially in the laboratory (Schnaufer et al. Reference Schnaufer, Domingo and Stuart2002). However, the discovery in laboratory generated-dyskinetoplastic T. b. brucei of a compensating mutation in the nuclear genome-encoded γ-subunit of the mitochondrial ATP synthase (Dean et al. Reference Dean, Gould, Dewar and Schnaufer2013) meant that the kinetoplast has been resurrected as the potential drug target of diminazene. These dyskinetoplastic lines do indeed show significant in vitro resistance to diamidines (including diminazene aceturate) and phenanthridines (Gould and Schnaufer, Reference Gould and Schnaufer2014). Furamidine (DB75), a closely related diamidine, whose fluorescent properties enabled tracking of its cellular distribution, was shown to bind to T. b. brucei kDNA and nuclear DNA in situ, and also to accumulate in other organelles identified as acidocalcisomes (Mathis et al. Reference Mathis, Holman, Sturk, Ismail, Boykin, Tidwell and Hall2006). The compound was also shown to interfere with the mitochondrial membrane potential (Lanteri et al. Reference Lanteri, Tidwell and Meshnick2008). Interestingly, it has been suggested that diminazene can also modulate the host immune response by dampening pro-inflammatory cytokines and excessive immune activation, which might also influence the in vivo effects of the drug (Kuriakose et al. Reference Kuriakose, Muleme, Onyilagha, Singh, Jia and Uzonna2012).
Chemically, diminazene is an aromatic diamidine made of two benzamidine moieties linked by a triazene bridge. Due to its charged nature, diminazene can only cross membranes via specific carriers and this has three important consequences: (a) the drug is not active on CNS infections as it cannot cross the blood–brain barrier; (b) the compound is selectively toxic to trypanosomes, as they express transporters that specifically accumulate diminazene; and (c) trypanosomes may become resistant to the drug by losing these transporters or their activity. As mentioned above, diminazene uptake in T. brucei mainly occurs via an aminopurine transporter called P2 or TbAT1, which is also implicated in the uptake of the related diamidine pentamidine and the melaminophenyl arsenical melarsoprol, two drugs licensed for HAT (Carter et al. Reference Carter, Berger and Fairlamb1995; Barrett and Fairlamb, Reference Barrett and Fairlamb1999; De Koning, Reference De Koning2008). Diminazene uptake into T. brucei is fast, with a Km of 0·45 µ m and a V max of 0·049 pm 107 cells−1 s−1 (De Koning et al. Reference De Koning, Anderson, Stewart, Burchmore, Wallace and Barrett2004) and is inhibited by pentamidine and adenosine, the main physiological substrate of this carrier. Loss of P2/TbAT1 activity was shown to cause diminazene resistance in T. b. brucei (Matovu et al. Reference Matovu, Stewart, Geiser, Brun, Maser, Wallace, Burchmore, Enyaru, Barrett, Kaminsky, Seebeck and de Koning2003), T. b. equiperdum (Barrett et al. Reference Barrett, Zhang, Denise, Giroud and Baltz1995; Stewart et al. Reference Stewart, Burchmore, Clucas, Hertz-Fowler, Brooks, Tait, MacLeod, Turner, de Koning, Wong and Barrett2010) and T. b. evansi (Witola et al. Reference Witola, Inoue, Ohashi and Onuma2004). Another gene, named TeDR40, has also been implicated in resistance in T. b. evansi (Witola et al. Reference Witola, Tsuda, Inoue, Ohashi and Onuma2005). However, using that gene to search for orthologues in other trypanosomatids at the TriTrypDB database (www.tritrypdb.org), indicates that it is actually a variant surface glycoprotein (VSG) gene, part of the parasite's system of antigenic variation whereby it avoids host immunity. It is possible that, in the process of selection of resistance, the parasites switched expression of a VSG gene independently of the resistance selection, which explains the massive increase in expression of that gene.
The application to T. brucei of a genome-wide RNA interference target sequencing (RIT-seq) screen, where any gene whose loss of function is identified by reduced drug sensitivity, was able to identify additional plasma membrane proteins (P-type H+-ATPases), as well as a putative protein phosphatase, that were linked to the action of the related diamidine pentamidine (Alsford et al. Reference Alsford, Eckert, Baker, Glover, Sanchez-Flores, Leung, Turner, Field, Berriman and Horn2012). The HAPT1/TbAQP2 carrier (De Koning, Reference De Koning2001b ), encoded by the TbAQP2 gene (Baker et al. Reference Baker, Glover, Munday, Aguinaga, Barrett, de Koning and Horn2012), has a key role in uptake of pentamidine and the melaminophenyl arsenicals in T. brucei, although its role in diminazene uptake is less pronounced (Teka et al. Reference Teka, Kazibwe, El-Sabbagh, Al-Salabi, Ward, Eze, Munday, Maser, Matovu, Barrett and de Koning2011; Munday et al. Reference Munday, Eze, Baker, Glover, Clucas, Aguinaga, Natto, Teka, McDonald, Lee, Graf, Ludin, Burchmore, Turner, Tait, MacLeod, Maser, Barrett, Horn and de Koning2014) and loss of P2/TbAT1 alone is sufficient to give high level of resistance to this latter drug (Matovu et al. Reference Matovu, Stewart, Geiser, Brun, Maser, Wallace, Burchmore, Enyaru, Barrett, Kaminsky, Seebeck and de Koning2003). It has recently been proposed that TbAQP2 acts as a receptor for pentamidine, with high affinity, and its uptake then occurs via receptor-mediated endocytosis (Song et al. Reference Song, Baker, Rothert, Henke, Jeacock, Horn and Beitz2016); further work is needed to confirm or refute this hypothesis, although other evidence points to pentamidine actually entering through the channel, enabled by a unique selectivity filter and the high degree of flexibility of the pentamidine chain (Munday et al. Reference Munday, Eze, Baker, Glover, Clucas, Aguinaga, Natto, Teka, McDonald, Lee, Graf, Ludin, Burchmore, Turner, Tait, MacLeod, Maser, Barrett, Horn and de Koning2014, Reference Munday, Settimo and de Koning2015a ).
Trypanosoma congolense appears to lack a functional equivalent of TbAQP2. A putative P2/TbAT1-type transporter, TcoAT1, was identified in T. congolense and a particular allele proposed to be associated with diminazene resistance (Delespaux et al. Reference Delespaux, Chitanga, Geysen, Goethals, Van den Bossche and Geerts2006). This conclusion was curious, given that the so-called resistance allele was not always associated with resistant form parasites isolated in one region (Delespaux et al. Reference Delespaux, Chitanga, Geysen, Goethals, Van den Bossche and Geerts2006) and was also abundant in areas where diminazene had not been used (Chitanga et al. Reference Chitanga, Marcotty, Namangala, Van den Bossche, Van Den Abbeele and Delespaux2011). Furthermore, TcoAT1 is not the orthologue of TbAT1, instead corresponding to a related, but distinct, member of the nucleoside transporter family (Munday et al. Reference Munday, Rojas Lopez, Eze, Delespaux, Van Den Abbeele, Rowan, Barrett, Morrison and de Koning2013). Its heterologous expression has proven that the encoded protein does not enable diminazene uptake, instead facilitating the uptake of adenosine and inosine (Munday et al. Reference Munday, Rojas Lopez, Eze, Delespaux, Van Den Abbeele, Rowan, Barrett, Morrison and de Koning2013). Hence, it can be definitively ruled out that the gene misnamed TcoAT1 has any role in diminazene uptake, action, or resistance.
Diminazene resistance is generally believed to be difficult to produce experimentally in T. congolense (in contrast to T. brucei). High levels of resistance to the drug were obtained in mice infected with T. b. evansi, but only when using immunocompromised animals, a result which stresses the importance of the link between immunity and chemotherapy, as the efficacy of trypanocides appears to be reduced by immunosuppression, hence favouring development of resistance (Osman et al. Reference Osman, Jennings and Holmes1992). In vitro experiments with T. b. brucei and T. b. evansi demonstrated that a shared mechanism of internalization accounts for the cross-resistance between diminazene and other diamidines as well as melaminophenyl arsenicals (melarsoprol and melarsomine) (Matovu et al. Reference Matovu, Stewart, Geiser, Brun, Maser, Wallace, Burchmore, Enyaru, Barrett, Kaminsky, Seebeck and de Koning2003). By contrast, no cross-resistance was observed with other chemically unrelated compounds including suramin or quinapyramine. A degree of cross-resistance has been observed between isometamidium and diminazene in T. brucei group trypanosomes, although the functional basis of this is not clear (Zhang et al. Reference Zhang, Giroud and Baltz1991; Witola et al. Reference Witola, Inoue, Ohashi and Onuma2004).
Homidium salts
Homidium bromide or ethidium bromide, also available as a chloride salt (Novidium®, Table 1), was introduced for field use in 1952, as an improvement to previous phenanthridine-based trypanocidal agents (Wainwright, Reference Wainwright2010). It is widely used in Africa to treat T. congolense and T. vivax infections in cattle, sheep and goats, in spite of its proven mutagenic and possible carcinogenic properties as a DNA intercalator (Sutcliffe et al. Reference Sutcliffe, Skellern, Araya, Cannavan, Sasanya, Dungu, van Gool, Munstermann and Mattioli2014). Due to its potential toxicity, the use of homidium is today highly discouraged (Sutcliffe et al. Reference Sutcliffe, Skellern, Araya, Cannavan, Sasanya, Dungu, van Gool, Munstermann and Mattioli2014). Widespread resistance to the drug in the 1960s and 1970s reduced its usage. Today, the number of doses of homidium used annually is reported to be down to around 10% of the total African trypanocide market, but this value may be a significant underestimate of its real use (Frans van Gool, personal communication, 2015).
Although used as a curative drug, homidium also possesses chemoprophylactic properties, but these are less pronounced than those of isometamidium (see subsection Isometamidium chloride below). For both purposes, homidium is administered at the dose of 1 mg kg−1 by a single, deep intramuscular injection (Peregrine, Reference Peregrine1994). Homidium excretion is faster than isometamidium, its serum concentration declining rapidly over the first 24 h following both intravenous and intramuscular injection at a standard dosage (Murilla et al. Reference Murilla, Holmes, Peregrine, Eisler and Ndung'u1999). Elimination half-life ranged from 178 h in Boran cattle to 488 h in Friesian cattle following intramuscular injection (Murilla et al. Reference Murilla, Holmes, Peregrine, Eisler and Ndung'u1999). However, low levels of the drug (0·1–0·3 ng mL−1) do persist in circulation for several weeks when given intramuscularly, providing an 8–17-week prophylaxis period (Dolan et al. Reference Dolan, Okech, Alushula, Mutugi, Stevenson, Sayer and Njogu1990; Murilla et al. Reference Murilla, Holmes, Peregrine, Eisler and Ndung'u1999). Homidium has an extensive extravascular distribution and accumulates predominantly in the liver and the kidneys (Murilla et al. Reference Murilla, Mdachi, Ismail and Karanja1996), a factor which presents some risk in products from treated animals destined for human consumption. Homidium can be used as sanative pair with diminazene, but not with isometamidium, where the shared phenanthridine core underlies cross-resistance (Peregrine et al. Reference Peregrine, Gray and Moloo1997).
Intracellular localization of homidium can be monitored by microscopy, exploiting the intrinsic fluorescence of the compound. Work on T. brucei showed that homidium localizes in the nucleus and the kinetoplast of treated trypanosomes (Cox et al. Reference Cox, Yielding and Yielding1984; Boibessot et al. Reference Boibessot, Turner, Watson, Goldie, Connel, McIntosh, Grant and Skellern2002). Treatment with the drug induces dyskinetoplasty in a similar way to other phenanthridines and diamidines (Riou et al. Reference Riou, Belnat and Benard1980; Shapiro and Englund, Reference Shapiro and Englund1990) and disruption of genome function has long been believed to underlie its trypanocidal effects. Indeed, it was found that homidium blocks both kinetoplast and nuclear DNA replication in T. brucei by distorting and changing the double helix topology (Roy Chowdhury et al. Reference Roy Chowdhury, Bakshi, Wang, Yildirir, Liu, Pappas-Brown, Tolun, Griffith, Shapiro, Jensen and Englund2010). The inhibition of minicircle replication and, consequently, loss of the kinetoplast network, was found to be the primary killing mechanism at low doses (0·02 µg mL−1), but at higher doses homidium was also shown to affect nuclear DNA, which could account for its ability to kill dyskinetoplastic trypanosomes (Roy Chowdhury et al. Reference Roy Chowdhury, Bakshi, Wang, Yildirir, Liu, Pappas-Brown, Tolun, Griffith, Shapiro, Jensen and Englund2010). The reason for the initial targeting of the kinetoplast over the nucleus is believed to be the result of the preferential accumulation of lipophilic cations (such as homidium) in the mitochondrion, as shown with other experimental trypanocides (Lanteri et al. Reference Lanteri, Tidwell and Meshnick2008; Ibrahim et al. Reference Ibrahim, Al-Salabi, El Sabbagh, Quashie, Alkhaldi, Escale, Smith, Vial and de Koning2011; Alkhaldi et al. Reference Alkhaldi, Martinek, Panicucci, Dardonville, Zikova and de Koning2016). The mechanism of resistance to homidium is not known, but it is likely to be similar to that of the related compound isometamidium.
Isometamidium chloride
Isometamidium chloride hydrochloride is a hybrid phenanthridine with amphiphilic and cationic properties, synthesized by coupling homidium with the diazotized p-aminobenzamide moiety of diminazene, modified with the amidine group in the meta position (see Table 1 for structures). It has both curative and prophylactic properties and, since its launch in the 1960s, it has remained the only drug available for chemoprophylaxis of AAT, after quinapyramine was discontinued due to problems linked to toxicity and, particularly, the induction of multi-drug resistance (Peregrine, Reference Peregrine1994; Geerts and Holmes, Reference Geerts and Holmes1998). The veterinary formulations are typically a mixture of four phenanthridine compounds: isometamidium chloride hydrochloride [8-(3-m-amidinophenyl-2-triazeno)-3-amino-5-ethyl-6-phenylphenanthridinium chloride hydrochloride], the positional red isomer [3-(3-m-amidinophenyl-2-triazeno)-8-amino-5-ethyl-6-phenylphenanthridinium chloride hydrochloride], the blue isomer [7-(m-amidinophenyldiazo)-3,8-diamino-5-ethyl-6-phenylphenanthridinium chloride hydrochloride], and the disubstituted compound [3,8-di(3-m-amidinophenyltriazeno)-5-ethyl-6-phenylphenanthridinium chloride dihydrochloride]. A protocol for their individual purification from the mixture and a detailed structural analysis of each compound were described in a recent publication (Igoli et al. Reference Igoli, Blackburn, Gray, Sutcliffe, Watson, Euerby and Skellern2015). In commercial products isometamidium is the principal component (guidelines establish it must be at least 55% of the total material), with the other components accounting for less than 40% (Sutcliffe et al. Reference Sutcliffe, Skellern, Araya, Cannavan, Sasanya, Dungu, van Gool, Munstermann and Mattioli2014). As the in vitro and in vivo trypanocidal activity on T. congolense is lower for the red and blue isomer it is paramount that the product composition follows strict quality standards (Sahin et al. Reference Sahin, Asencio, Izotte, Pillay, Coustou, Karembe and Baltz2014). The disubstituted compound has poor trypanocidal activity but it has a good prophylactic effect, possibly because it can act as a pro-drug that is cleaved to isometamidium in vivo (Sahin et al. Reference Sahin, Asencio, Izotte, Pillay, Coustou, Karembe and Baltz2014).
Isometamidium is used primarily to treat and prevent T. congolense and T. vivax infections in livestock in Africa. Its activity against T. brucei spp. is less marked, but this drug can also be utilized against some T. b. evansi strains, although not when these have reached the CNS, as the compound does not cross the blood–brain barrier. The drug is administered to cattle at single doses of 0·25–1·0 mg kg−1 for cure, and at doses of 0·5–1 mg kg−1 for prophylaxis (Leach and Roberts, Reference Leach and Roberts1981). The dosage for T. b. evansi infections is generally 1–2 mg kg−1, but in horses it is recommended not to exceed 0·5 mg kg−1 due to toxicity issues (Uilenberg, Reference Uilenberg1998; Desquesnes et al. Reference Desquesnes, Dargantes, Lai, Lun, Holzmuller and Jittapalapong2013a ). Multiple intramuscular administrations of isometamidium can cause severe fibrous lesions, hence damaging the carcass and meat quality from livestock. Intravenous administration has been successfully used to abrogate muscular damage, but it has been suggested that this could result in compromised prophylactic activity, due to the lack of a drug depot at the injection site (Dowler et al. Reference Dowler, Schillinger and Connor1989; Munstermann et al. Reference Munstermann, Mbura, Maloo and Lohr1992). The duration of prophylactic activity following intramuscular administration in cattle is typically 2–3 months and may be up to 6 months, but can vary greatly, depending on the formulation and dosage used and on the parasite strain, as well as on other factors, including susceptibility of the particular breed and its general health status (Toro et al. Reference Toro, Leon, Lopez, Pallota, Garcia and Ruiz1983; Kinabo and Bogan, Reference Kinabo and Bogan1988).
Isometamidium plasma concentrations reach their peak within 1 h after administration and then fall relatively quickly during the first week post-treatment and thereafter more gradually (Kinabo, Reference Kinabo1993; Eisler et al. Reference Eisler, Arowolo, Gault, Moloo, Holmes and Peregrine1994). Three months after cattle had been injected, the circulating drug concentration was measured at 0·75 ng mL−1 (Eisler et al. Reference Eisler, Arowolo, Gault, Moloo, Holmes and Peregrine1994). This study showed that the serum concentration fits a bi-exponential model, with half-life of approximately 25 days for the second phase in cattle (Eisler et al. Reference Eisler, Arowolo, Gault, Moloo, Holmes and Peregrine1994), while another study (Eisler, Reference Eisler1996) indicated an elimination half-life of 9–19 days. In sheep and goats isometamidium appears to be eliminated more rapidly than in cattle (Wesongah et al. Reference Wesongah, Jones, Kibugu and Murilla2004). The drug accumulates in the liver, kidneys and spleen as well as at the injection site, and from here it is slowly released to the plasma exerting its prophylactic activity (Kinabo and Bogan, Reference Kinabo and Bogan1988). Persistence of isometamidium residues is much longer than for diminazene. For this reason, a withdrawal period of 30 days was established for consumption of produce from cattle treated with the drug (FAO, 1990), although in practice the withdrawal (discard) period is always product-specific. Excretion occurs mainly via bile and levels in cattle milk are generally very low (Kinabo, Reference Kinabo1993).
Isometamidium may be used as part of a sanative pair with diminazene, the two drugs being used sequentially to minimize the risk of resistance development (Leach and Roberts, Reference Leach and Roberts1981; Peregrine, Reference Peregrine1994). Despite this recommendation, there are multiple reports of field isolates, from many African countries, indicating isometamidium resistance, particularly in T. congolense but also in T. brucei species and T. vivax, sometimes detailing cross-resistance with diminazene (Ainanshe et al. Reference Ainanshe, Jennings and Holmes1992; Clausen et al. Reference Clausen, Sidibe, Kabore and Bauer1992; Codjia et al. Reference Codjia, Mulatu, Majiwa, Leak, Rowlands, Authie, d'Ieteren and Peregrine1993; Afewerk et al. Reference Afewerk, Clausen, Abebe, Tilahun and Mehlitz2000; Sinyangwe et al. Reference Sinyangwe, Delespaux, Brandt, Geerts, Mubanga, Machila, Holmes and Eisler2004; Mamoudou et al. Reference Mamoudou, Delespaux, Chepnda, Hachimou, Andrikaye, Zoli and Geerts2008). However, other reports found no cross-resistance (e.g. Gray et al. Reference Gray, Kimarua, Peregrine and Stevenson1993; Joshua et al. Reference Joshua, Obwolo, Bwangamoi and Mandebvu1995) and we conclude that cross-resistance does not necessarily occur, but may be a consequence of the level of resistance that has been established, whereas in other cases resistance to both drugs may have been induced separately. In addition, the chance of cross-resistance developing may be different for the various animal trypanosome species, given their known differences in biochemical physiology and drug transport.
By taking advantage of isometamidium's intrinsic fluorescence, accumulation in the kinetoplast was observed (Wilkes et al. Reference Wilkes, Peregrine and Zilberstein1995; Boibessot et al. Reference Boibessot, Turner, Watson, Goldie, Connel, McIntosh, Grant and Skellern2002). Although closely related to the intercalating phenanthridine homidium, isometamidium is not known to be carcinogenic, and was reported to bind kDNA with an unconventional ‘sideways’ geometry (Dougherty and Waring, Reference Dougherty and Waring1982). Its high affinity for the kDNA might underlie its trypanocidal activity. Linearization of kDNA minicircles in T. b. equiperdum following interaction of the drug with the kinetoplast was observed (Shapiro and Englund, Reference Shapiro and Englund1990). Moreover, naturally occurring dyskinetoplastic T. b. evansi (Brun and Lun, Reference Brun and Lun1994) and in vitro-generated T. b. brucei lacking a functional kinetoplast (Gould and Schnaufer, Reference Gould and Schnaufer2014) are highly resistant to the drug. Efficacy against some T. b. evansi strains might relate to these parasites retaining kDNA (albeit dispersed in dyskinetoplastidy) while others are akinetoplastic (i.e. retain no kDNA at all) and may be less susceptible to the drug. However, the drug would still accumulate preferentially in the mitochondrion, as the mitochondrial membrane potential is unaffected by the loss of the kinetoplast in cells carrying a compensatory mutation in the γ-subunit of the F1F0-ATP synthase (Dean et al. Reference Dean, Gould, Dewar and Schnaufer2013), providing a driving force for cations. A mutation in this ATP synthase subunit is sufficient to cause a substantial level of isometamidium and homidium resistance, although further drug pressure was shown to increase this even further. Interestingly, this very high level of resistance is indeed associated with a loss of mitochondrial membrane potential, preventing further isometamidium accumulation in this organelle (Eze et al. Reference Eze, Gould, Munday, Tagoe, Stelmanis, Schnaufer and de Koning2016).
Despite possessing the recognition motif for the P2/TbAT1 transporter and despite being a high-affinity inhibitor of this carrier (De Koning, Reference De Koning2001a ), the internalization of isometamidium depends at most partially on this route (Delespaux and de Koning, Reference Delespaux and de Koning2007). Passive diffusion across the membrane may be feasible but is not likely, given the two positive charges on the molecule and partial characterization of isometamidium transport, linking drug resistance, at least in part, to reduced uptake (Sutherland et al. Reference Sutherland, Mounsey and Holmes1992; Wilkes et al. Reference Wilkes, Peregrine and Zilberstein1995, Reference Wilkes, Mulugeta, Wells and Peregrine1997). High-throughput RIT-seq (Baker et al. Reference Baker, Hamilton, Wilkes, Hutchinson, Barrett and Horn2015) failed to identify involvement of any of the receptor-mediated endocytosis pathways as previously identified for suramin (see subsection Suramin sodium below) using this approach (Alsford et al. Reference Alsford, Eckert, Baker, Glover, Sanchez-Flores, Leung, Turner, Field, Berriman and Horn2012), although alternative endocytic routes could not be ruled out.
Resistance to isometamidium is encountered in the field. In T. congolense a mechanism behind resistance was proposed to relate to diminished mitochondrial membrane potential (Wilkes et al. Reference Wilkes, Mulugeta, Wells and Peregrine1997). This, in turn, would diminish the accumulation of drug in the mitochondrion, having a net effect of reduced uptake at the plasma membrane, presumably due to rapid equilibration of intracellular and extracellular concentrations when the mitochondrial sink is lost. Active extrusion by plasma membrane transporters has also been proposed (Sutherland and Holmes, Reference Sutherland and Holmes1993). A recent application of the RIT-seq approach, conducted on T. brucei, identified mutations to many subunits of the vacuolar ATPase (found in the lysosomes and acidocalcisomes), in the trafficking protein AP-3 (an adaptin that mediates delivery of proteins to lysosome-related organelles) and in EMC (an ER membrane complex) that reduced drug activity, potentially contributing to dug resistance (Baker et al. Reference Baker, Hamilton, Wilkes, Hutchinson, Barrett and Horn2015). Secondary loss of kDNA was found to be possible once vATPase and AP-3 subunits are lost from the cells, pointing to an intriguing, but as yet ill-defined, interaction between the vacuolar system and mitochondrion. The fact that kDNA is lost in cells selected for resistance to isometamidium was classically interpreted to point to its role as target. However, the discovery that kDNA loss can occur as a consequence of changes to the vacuolar system complicates this interpretation.
Quinapyramine sulphate
Quinapyramine sulphate was developed from the early trypanocide Surfen C (Curd and Davey, Reference Curd and Davey1950) and came into use around 1950. The compound was applied to treat cattle infected with trypanosomes until 1976, when it was withdrawn from many areas due to emergence of widespread resistance (Connor, Reference Connor1992). The drug was subsequently reintroduced in 1984 to treat T. b. evansi in camels and horses (Peregrine, Reference Peregrine1994), and is still used today (Ranjithkumar et al. Reference Ranjithkumar, Saravanan, Yadav, Kumar, Singh and Dey2014). In horses with acute infections of T. brucei spp. quinapyramine is considered the most effective treatment (5 mg kg−1 via subcutaneous injection), although the drug induces severe but transient side effects in these animals (Auty et al. Reference Auty, Mundy, Fyumagwa, Picozzi, Welburn and Hoare2008). The prosalt form of quinapyramine (a mixture of the soluble sulphate and the insoluble chloride salts) was the first prophylactic drug available for animal infections. A 7·4 mg kg−1 dose of this prosalt suspension has both a curative and a prophylactic (up to 4 months) effect on T. b. evansi infections in horses and camels (Williamson, Reference Williamson and Mulligan1970).
Quinapyramine is a quinoline pyrimidine (Table 1) and, as isometamidium and diminazene, a dication at physiological pH (homidium is monocationic). As seen for the other charged trypanocides, quinapyramine is unable to cross the blood-brain barrier, which explains its failure to cure T. b. evansi infections in equids when the CNS is affected (Ranjithkumar et al. Reference Ranjithkumar, Saravanan, Yadav, Kumar, Singh and Dey2014). However, it is important to note that some cationic trypanocides do penetrate the blood–brain barrier, the clearest example being compound DB829 (Wenzler et al. Reference Wenzler, Yang, Braissant, Boykin, Brun and Wang2013). Pentamidine has actually been used to treat ‘early-late stage’ HAT (Doua et al. Reference Doua, Miezan, Sanon Singaro, Boa Yapo and Baltz1996) but its movement across the blood–brain barrier is counteracted by active efflux mechanisms, including P-glycoprotein and multi-drug resistance transporters (Sanderson et al. Reference Sanderson, Dogruel, Rodgers, De Koning and Thomas2009).
Plasma levels of quinapyramine decline rapidly after dosing and, in the case of the prosalt, its persistence is probably due to slow release from the subcutaneous depot formed at the injection site (Spinks, Reference Spinks1950). Quinapyramine accumulates in the liver and kidneys, where its concentration remains high for weeks and can cause organ-specific toxicity. Excretion occurs mainly via urine (Spinks, Reference Spinks1950).
Quinapyramine's mode of action remains unknown. Hypotheses include the interference with nucleic acid synthesis and inhibition of cytoplasmic ribosomes (and, therefore, protein synthesis) (Newton, Reference Newton1962, Reference Newton1966). However, its dicationic/aromatic nature would strongly suggest a mitochondrial accumulation, as with the phenanthridines and bis-benzamidines.
Trypanosoma congolense and T. b. evansi lines resistant to the drug can easily be obtained by in vivo selection in mice (Ndoutamia et al. Reference Ndoutamia, Moloo, Murphy and Peregrine1993; Liao and Shen, Reference Liao and Shen2010). As quinapyramine resistant T. congolense trypanosomes show cross-resistance to isometamidium, homidium and diminazene, the use of this compound to treat infections in cattle is not recommended (Peregrine et al. Reference Peregrine, Gray and Moloo1997). Given the lack of cross-resistance between diminazene and homidium, the fact that quinapyramine is cross-resistant to both is intriguing. Although the mechanism underpinning quinapyramine resistance remains unknown, it is likely that all these trypanocides have a mitochondrial target and that any single change that dramatically reduces the mitochondrial membrane potential, or the loss of organic cation carriers in the inner mitochondrial membrane, could result in resistance to all of them.
Suramin sodium
Suramin sodium is a symmetrical polyanionic sulfonated naphthylamine (Table 1). It is the oldest trypanocide still in use, having been introduced in 1921 for the treatment of surra in camels and replacing the then-standard treatment of intravenous tartar emetic (potassium antimonyl tartrate) (Uilenberg, Reference Uilenberg1998). A single dose of 6–10 g of suramin sodium per camel was described as 100% effective (Bennett, Reference Bennett1930). Suramin is also the standard treatment for equine trypanosomiasis (T. brucei spp.), being more effective than diminazene and less toxic than quinapyramine (Williamson, Reference Williamson and Mulligan1970). The current treatment for camels and horses is 10 mg kg−1, administered intravenously. Intramuscular administration is avoided as it causes intense local irritation. Suramin has further been used for cure and prophylaxis of onchocerciasis and other microfilarial infections including Brugia pahangi (Delespaux and de Koning, Reference Delespaux and de Koning2007), as well as for the treatment of early stage HAT since 1922 (Apted, Reference Apted and Mulligan1970). Although suramin is effective against T. b. gambiense (Knobloch et al. Reference Knobloch, Tischendorf, Konig and Mehlitz1984; Pepin and Khonde, Reference Pepin and Khonde1996), it is mostly used against HAT due to T. b. rhodesiense, for which it is still available today (Voogd et al. Reference Voogd, Vansterkenburg, Wilting and Janssen1993), whereas it was replaced with pentamidine for the form due to T. b. gambiense. Although the drug has good efficacy against T. simiae in pigs (Stephen, Reference Stephen1966; Williamson, Reference Williamson and Mulligan1970), it is relatively ineffective against T. congolense and T. vivax (Leach and Roberts, Reference Leach and Roberts1981), presumably due to the aforementioned differences in biochemical physiology that distinguish T. brucei group organisms from these other species.
Old work showed that suramin can be used as a prophylactic agent when administered subcutaneously as an insoluble complex with one of the cationic trypanocides (e.g. with quinapyramine, in a 1:3 molecular proportion, reflecting the six negative charges of suramin vs the two cationic charges of quinapyramine), resulting in 3–6 months protection at 40 mg kg−1 of quinapyramine in pigs (Williamson, Reference Williamson and Mulligan1970) and >160 days protection in cattle (Williamson and Desowitz, Reference Williamson and Desowitz1956). This approach could be effective for the eradication of T. b. gambiense in pigs, which are reportedly acting as reservoir hosts of this species (Mehlitz et al. Reference Mehlitz, Zillmann, Scott and Godfrey1982). Complexes of suramin with homidium, quinapyramine and prothidium also gave protection in experimental infections in cattle (Desowitz, Reference Desowitz1957).
The pharmacokinetic parameters of suramin in animals (Kinabo, Reference Kinabo1993) have not been subject to the same extensive characterization as occurred in humans, where the compound has also been trialled for the treatment of AIDS and cancer (Barrett et al. Reference Barrett, Boykin, Brun and Tidwell2007). Most of the drug (>99%) binds to plasma proteins yielding a slow clearance. The terminal half-life in humans ranges between 40 and 50 days or more, depending on the infusion protocol applied (Jodrell et al. Reference Jodrell, Reyno, Sridhara, Eisenberger, Tkaczuk, Zuhowski, Sinibaldi, Novak and Egorin1994). This slow clearance underpins limited (i.e. several weeks) prophylactic action in animals too when the drug is used on its own. Suramin does suppress infection, but is dependent on the host's immune response to be fully effective (Leach and Roberts, Reference Leach and Roberts1981). Because of its large molecular size and highly anionic nature, suramin does not cross the blood–brain barrier.
Suramin strongly binds to human serum proteins and various trypanosome enzymes by electrostatic interaction (Voogd et al. Reference Voogd, Vansterkenburg, Wilting and Janssen1993). The drug was proposed to enter trypanosomes via receptor-mediated uptake bound to LDL and to accumulate in the lysosome (Vansterkenburg et al. Reference Vansterkenburg, Coppens, Wilting, Bos, Fischer, Janssen and Opperdoes1993). This hypothesis, however, looked doubtful after it was demonstrated that in T. brucei (procyclic form at least) suramin and LDL uptake are not coupled (Pal et al. Reference Pal, Hall and Field2002). A definitive mode of action for the compound has not been determined. Fairlamb and Bowman proposed that suramin curbs glycolytic ATP production in T. brucei by inhibiting glycerol-3-phospate oxidase and NAD+-dependent glycerol-3-phosphate dehydrogenase (Fairlamb and Bowman, Reference Fairlamb and Bowman1980). However, being highly charged, suramin binds many enzymes when assayed and a multitude of putative targets have been proposed (Gutteridge, Reference Gutteridge1985), including 6-phosphogluconate dehydrogenase, of the pentose phosphate pathway, of which it is a competitive inhibitor (Hanau et al. Reference Hanau, Rippa, Bertelli, Dallocchio and Barrett1996). More recently, a RIT-seq screen in bloodstream T. brucei identified 28 genes that contribute to suramin action, including: a surface glycoprotein family (ISG75), which appears to be the ligand to which the drug binds; cathepsin L, believed to release the drug from ligand within the lysosomal system; a number of deubiquitinating enzymes and various proteins involved in the endocytic pathway (Alsford et al. Reference Alsford, Eckert, Baker, Glover, Sanchez-Flores, Leung, Turner, Field, Berriman and Horn2012). It appears that inhibiting uptake of suramin, or its normal passage through the endocytic pathway following binding to a specific receptor, is sufficient to render parasites resistant to the drug, although it remains unknown how suramin kills once accumulated intracellularly.
Extensive use of the compound in the first half of the 20th century resulted in emergence of widespread resistance in T. b. evansi in Africa (Boid et al. Reference Boid, Jones and Payne1989; El Rayah et al. Reference El Rayah, Kaminsky, Schmid and El Malik1999) and South-East Asia (Gill, Reference Gill1971; Zhou et al. Reference Zhou, Shen, Liao, Zhou and Lin2004), in some cases leading to withdrawal of suramin as a treatment (El Rayah et al. Reference El Rayah, Kaminsky, Schmid and El Malik1999). However, even in the absence of drug pressure, the resistance phenotype has persisted in the field, as found for some Sudanese T. b. evansi strains (El Rayah et al. Reference El Rayah, Kaminsky, Schmid and El Malik1999). Stability of the suramin resistance phenotype was also observed in T. brucei lines generated in vitro (Scott et al. Reference Scott, Tait and Turner1996) and in T. b. evansi parasites selected in mice (Mutugi et al. Reference Mutugi, Boid and Luckins1994). However, the drug was effective against T. b. evansi isolates in Brazil, where it had not been used (Faccio et al. Reference Faccio, Da Silva, Gressler, Tonin, Lazzarotto, Miletti and Monteiro2013).
Melarsomine dihydrochloride
An early reported case of an attempt to cure an animal afflicted with trypanosomiasis was that of Dr David Livingstone, the Scottish missionary whose travels in Southern Africa in the mid-19th century were exceptionally well recorded. In a letter to the British Medical Journal in 1858 he described the use of arsenic oxide (Fowler's solution) to treat a case of ‘fly disease’ in a horse (Livingstone, Reference Livingstone1858). Although the treated horse was not cured, there was a temporary relief in symptoms. Over 50 years later, once the trypanosome had been implicated, H. W. Thomas and A. Breinl, and then P. Ehrlich, revisited arsenic chemistry to seek trypanocides in the early days of chemotherapy (Williamson, Reference Williamson and Mulligan1970). By the 1950s melarsoprol had been introduced for the treatment of late-stage HAT; the drug was created by coupling of melarsen oxide to 2,3-dimercaptopropanol (Steverding, Reference Steverding2010). The formulation displayed diminished toxicity while retaining potent trypanocidal activity.
Melarsomine dihydrochloride (Table 1) is a melamino-phenylarsine, synthesized by linking melarsen oxide (Barrett et al. Reference Barrett, Boykin, Brun and Tidwell2007) to two equivalent of cysteamine (Berger and Fairlamb, Reference Berger and Fairlamb1994). The compound has improved aqueous solubility over melarsoprol. It was introduced to the market in 1992 and is the latest addition to the veterinary trypanocidal list. The drug (Immiticide®) is also used in the treatment of heartworms in dogs, where it kills adult worms (McCall et al. Reference McCall, McTier, Dzimianski, Raynaud and Holmes1994), albeit with a low margin of safety. It is registered for use against T. b. evansi in camels at a dose of 0·25 mg kg−1, but it has also been evaluated and proven efficacious against T. b. evansi infections in cattle (Desquesnes et al. Reference Desquesnes, Kamyingkird, Vergne, Sarataphan, Pranee and Jittapalapong2011), goats (Gutierrez et al. Reference Gutierrez, Corbera, Bayou and van Gool2008) and horses (Tamarit et al. Reference Tamarit, Gutierrez, Arroyo, Jimenez, Zagala, Bosch, Sirvent, Alberola, Alonso and Caballero2010), although at higher dosages than that applied to camels (i.e. 0·5 mg kg−1 or above). Melarsomine also proved curative in cattle infected with T. b. evansi strains resistant to suramin (Payne et al. Reference Payne, Sukanto, Partoutomo and Jones1994). Moreover, treatment regimens with both 0·25 and 0·5 mg kg−1 of the drug were proven effective in curing acute and chronic T. b. equiperdum infections in horses, resulting in a reduction of neurological symptoms (Hagos et al. Reference Hagos, Goddeeris, Yilkal, Alemu, Fikru, Yacob, Feseha and Claes2010) and offering a possible treatment for these infections. Side effects to the drug are usually mild (salivation, lacrimation, muscle tremors, increased gut motility and frequent urination), but a severe adverse reaction has also been documented (Berlin et al. Reference Berlin, Nasereddin, Azmi, Ereqat, Abdeen and Baneth2010). Reports of neurological sequelae in dogs (Hettlich et al. Reference Hettlich, Ryan, Bergman, Marks, Lewis, Bahr, Coates, Mansell and Barton2003), albeit perhaps not analogous to the reactive encephalopathy associated with melarsoprol treatment of humans (Blum et al. Reference Blum, Nkunku and Burri2001), are notable. Should the reduced neurotoxicity of melarsomine be replicated in man it might be considered as a replacement for melarsoprol, although it is doubtful that comparative clinical trials of the two arsenicals would receive ethical clearance, especially since melarsoprol is being phased out in favour of nifurtimox–eflornithine combination therapy (Simarro et al. Reference Simarro, Franco, Diarra, Postigo and Jannin2012). The paucity of compounds that kill adult filarial worms is of note too, and, should the safety profile of melarsomine be acceptable, it could be considered for use against the human filariases.
The mode of action of melarsomine is unknown. As for other trypanocidal arsenicals, the disruption of the thiol-redox balance is a possible mechanism (Fairlamb, Reference Fairlamb2003). The drug (or, rather, its metabolite melarsen oxide) enters T. brucei via the same P2/TbAT1 adenosine nucleoside transporter (Carter and Fairlamb, Reference Carter and Fairlamb1993; De Koning and Jarvis, Reference De Koning and Jarvis1999) and TbAQP2 (Munday et al. Reference Munday, Eze, Baker, Glover, Clucas, Aguinaga, Natto, Teka, McDonald, Lee, Graf, Ludin, Burchmore, Turner, Tait, MacLeod, Maser, Barrett, Horn and de Koning2014) that carry other melaminophenyl arsenicals and the diamidine trypanocides. Selective uptake probably accounts for most of the selective toxicity of the arsenicals (Baker et al. Reference Baker, de Koning, Maser and Horn2013). Reduction of P2/TbAT1 activity is a known reason behind onset of cross-resistance between the compounds that enter via this route: trypanosomes of the T. brucei group resistant to melarsomine are often also less sensitive to diamidines and other arsenical drugs as melarsoprol, but not to suramin (Zhang et al. Reference Zhang, Giroud and Baltz1991; Pospichal et al. Reference Pospichal, Brun, Kaminsky and Jenni1994). In vitro and in vivo selected melarsomine-resistant T. b. evansi (Suswam et al. Reference Suswam, Taylor, Ross and Martin2001) revealed that the decrease in P2/TbAT1 transporter activity was linked both to reduced transporter expression and changes in binding properties (Suswam et al. Reference Suswam, Ross and Martin2003). In a T. b. brucei strain selected for melarsomine resistance in mice the TbAT1 gene was still present but its transcript was lost (Stewart et al. Reference Stewart, Burchmore, Clucas, Hertz-Fowler, Brooks, Tait, MacLeod, Turner, de Koning, Wong and Barrett2010). The lack of authentic orthologues of TbAT1 and TbAQP2 in T. congolense and T. vivax (see subsection Diminazene aceturate above) may explain why the drug is less potent against these parasites.
DRUG RESISTANCE IN THE FIELD: DEFINITION AND EXTENT OF THE PROBLEM
Drug resistance is suspected when treatment failure occurs using standard drug dosages. However, in the field, this interpretation can be erroneous, as treatment failure can result from many factors other than the parasite's increased tolerance to drugs. For example, the presence of parasites in treated animals could correspond to a new infection rather than to recrudescence, particularly in areas of high challenge (Rowlands et al. Reference Rowlands, Leak, Peregrine, Nagda, Mulatu and d'Ieteren2001). Using microsatellite DNA markers to strain type T. congolense from cattle in Ethiopia following treatment with diminazene, essentially equal occurrences of new infection (40%) and actual relapse (37·5%) were proposed (Moti et al. Reference Moti, De Deken, Thys, Van Den Abbeele, Duchateau and Delespaux2015). Other causes of treatment failure not linked to true drug resistance could be related to the poor health state of the animal (e.g. malnutrition, immunosuppression, concurrent infections), or to incorrect drug use (e.g. irregular treatment or prolonged intervals between treatments), or to under-dosage. The latter can result from poor drug quality (either due to inappropriate storage or to the use of counterfeit products) (Sutcliffe et al. Reference Sutcliffe, Skellern, Araya, Cannavan, Sasanya, Dungu, van Gool, Munstermann and Mattioli2014), or from incorrect drug usage (wrong dilution, use of unsterilized water or erroneous dosage due to inaccurate estimation of the animal weight) (Van den Bossche et al. Reference Van den Bossche and Delespaux2000; Grace et al. Reference Grace, Randolph, Affognon, Dramane, Diall and Clausen2009). For phenanthridines, in particular isometamidium, the adverse reaction which often appears at the injection site might possibly alter drug absorption and diminish the levels of drug in circulation (Kinabo, Reference Kinabo1993), thus determining under-dosage. It is widely believed that under-dosing could represent a major determinant in drug resistance development in the field through parasite exposure to sub-curative drug concentrations (Leach and Roberts, Reference Leach and Roberts1981). A similar phenomenon could derive from failure to comply with strict dose timing, which could lead to periods where sub-prophylactic drug levels are present (Leach and Roberts, Reference Leach and Roberts1981). Moreover, as a mutagen, homidium might also directly contribute to resistance appearance through induction of mutations in parasites that are then selected under drug pressure. Constant parasitological monitoring is necessary to distinguish treatment failure from appearance of true resistance.
In the previous section, we have outlined that there are issues associated with selected resistance to each of the drugs used against the animal trypanosomiases. Cases of resistance to veterinary trypanocides started to be reported in the field soon after their introduction, and their numbers have been increasing ever since (Delespaux et al. Reference Delespaux, Geysen, Van den Bossche and Geerts2008b ). A review of available literature in 2008 reported loss of efficacy of the available AAT trypanocides in at least 17 African countries (Delespaux et al. Reference Delespaux, Geysen, Van den Bossche and Geerts2008b ). Available data, in 2001, indicated that resistance to isometamidium was more widespread than resistance to diminazene (Geerts et al. Reference Geerts, Holmes, Eisler and Diall2001), however, this may no longer be so, as prevalence of resistance may change substantially over a few years (Delespaux et al. Reference Delespaux, Dinka, Masumu, Van den Bossche and Geerts2008a ). Treatment failure against T. congolense and T. vivax infections with either of these drugs has been observed in both West (Kupper and Wolters, Reference Kupper and Wolters1983; Pinder and Authie, Reference Pinder and Authie1984; Knoppe et al. Reference Knoppe, Bauer, McDermott, Peregrine, Mehlitz and Clausen2006; Mungube et al. Reference Mungube, Vitouley, Allegye-Cudjoe, Diall, Boucoum, Diarra, Sanogo, Randolph, Bauer, Zessin and Clausen2012; Vitouley et al. Reference Vitouley, Sidibe, Bengaly, Marcotty, Van Den Abbeele and Delespaux2012) and East Africa (Mbwambo et al. Reference Mbwambo, Mella and Lekaki1988; Chitambo and Arakawa, Reference Chitambo and Arakawa1992a ; Dagnachew et al. Reference Dagnachew, Terefe, Abebe, Barry, McCulloch and Goddeeris2015; Moti et al. Reference Moti, De Deken, Thys, Van Den Abbeele, Duchateau and Delespaux2015). More worryingly, strains of T. congolense resistant to both isometamidium and diminazene have been detected in several locations, including Cameroon (Mamoudou et al. Reference Mamoudou, Delespaux, Chepnda, Hachimou, Andrikaye, Zoli and Geerts2008), Burkina Faso (Clausen et al. Reference Clausen, Sidibe, Kabore and Bauer1992), Ethiopia (Codjia et al. Reference Codjia, Mulatu, Majiwa, Leak, Rowlands, Authie, d'Ieteren and Peregrine1993; Afewerk et al. Reference Afewerk, Clausen, Abebe, Tilahun and Mehlitz2000; Moti et al. Reference Moti, Fikru, Van Den Abbeele, Buscher, Van den Bossche, Duchateau and Delespaux2012), Somalia (Ainanshe et al. Reference Ainanshe, Jennings and Holmes1992) and Zambia (Sinyangwe et al. Reference Sinyangwe, Delespaux, Brandt, Geerts, Mubanga, Machila, Holmes and Eisler2004), rendering their use as a sanative pair inoperative. These multiple resistant stocks might be the result of separate selection processes for the two drugs, as cross-resistance between diminazene and isometamidium has been considered a rare phenomenon.
Drug resistance to animal trypanocides has also been reported from outside of Africa. For example, T. vivax strains refractory to diminazene were identified in South America, where the compound is the first line drug to treat these infections (Desquesnes et al. Reference Desquesnes, de La Rocque and Peregrine1995; Cadioli et al. Reference Cadioli, Barnabé, Machado, Teixeira, André, Sampaio, Fidélis Junior, Teixeira and Marques2012). Diminazene treatment failure against T. b. evansi infections in horses and mules in Thailand has also been reported, following decades of use (Tuntasuvan et al. Reference Tuntasuvan, Jarabrum, Viseshakul, Mohkaew, Borisutsuwan, Theeraphan and Kongkanjana2003). Trypanosoma b. evansi strains resistant to suramin (Zhou et al. Reference Zhou, Shen, Liao, Zhou and Lin2004) and quinapyramine (Zhou et al. Reference Zhou, Shen, Liao, Zhou and Lin2004; Liao and Shen, Reference Liao and Shen2010) have been reported in China as well as in Africa (El Rayah et al. Reference El Rayah, Kaminsky, Schmid and El Malik1999).
TESTS FOR RESISTANCE DETECTION
In vivo methods
Because of the confounding factors that can cause treatment failures, outlined above, methods to assess true resistance are crucial. However, reliable tests have been relatively difficult to establish for widespread use in settings where AAT is endemic. Methods such as the ‘block treatment’ approach (Delespaux et al. Reference Delespaux, Geysen, Van den Bossche and Geerts2008b ) have been proposed to enable identification of probable resistance in the field, whereby cattle in a particular location are split into control and treated groups and followed to first detection of parasitaemia. Broadly, the presence of resistance is measured by comparing time to parasite detection in treated vs the untreated controls (Eisler et al. Reference Eisler, McDermott, Mdachi, Murilla, Sinyangwe, Mubanga, Machila, Mbwambo, Coleman, Clausen, Bauer, Sidibé, Geerts, Holmes and Peregrine2000): the closer to the control group, the more likely the presence of resistance. However, although there are logistical advantages to such tests (e.g. no requirement for parasite isolation), they still require considerable investment in time and numbers of cattle involved (typically revisits every two weeks for 10–14 weeks, suggested group sizes of 30–80 animals), and the results are only indicative of resistance. Confirmatory trypanocide efficacy studies against veterinary trypanosomes still rely primarily on infection and treatment experiments in the natural hosts or in laboratory animals (i.e. rodents), where parasite clearance from blood following treatment is assessed by microscopy (Eisler et al. Reference Eisler, Brandt, Bauer, Clausen, Delespaux, Holmes, Ilemobade, Machila, Mbwambo, McDermott, Mehlitz, Murilla, Ndung'u, Peregrine, Sidibe, Sinyangwe and Geerts2001). However, the requirement for long follow-up periods (i.e. 100 days for in vivo tests in ruminants and 60 days when using mice models) makes tests cumbersome, expensive and slow, as well as susceptible to the confounding factor of re-infection after successful treatment when tests are undertaken in the field. Extrapolation of rodent data to ruminants is not necessarily an accurate reflection of treatment success in large animals and, crucially, neither T. vivax nor many T. congolense strains adapt readily to propagation in mice (Eisler et al. Reference Eisler, Brandt, Bauer, Clausen, Delespaux, Holmes, Ilemobade, Machila, Mbwambo, McDermott, Mehlitz, Murilla, Ndung'u, Peregrine, Sidibe, Sinyangwe and Geerts2001). Nevertheless, the single-dose mouse test is currently considered the standard test to study single or multiple resistance in T. congolense and T. brucei isolates at an accelerated rate. In spite of its being non-quantitative, the test does offer a relatively rapid (60 days) means to qualitatively assess whether parasites respond to doses of drug routinely used in veterinary practice or not. Substitution of microscopy with PCR techniques, such as the ITS1 TD PCR (Tran et al. Reference Tran, Napier, Rowan, Cordel, Labuschagne, Delespaux, Van Reet, Erasmus, Joubert and Buscher2014) for the detection of trypanosomes in blood, offers a mean to improve drug sensitivity studies using mouse models.
In vitro methods
Laboratory cultivation of bloodstream form T. brucei transformed our ability to assess sensitivity to drugs, especially in the quantities made possible by large chemical libraries and robotic screening, resulting in new lead compounds (Nare et al. Reference Nare, Wring, Bacchi, Beaudet, Bowling, Brun, Chen, Ding, Freund, Gaukel, Hussain, Jarnagin, Jenks, Kaiser, Mercer, Mejia, Noe, Orr, Parham, Plattner, Randolph, Rattendi, Rewerts, Sligar, Yarlett, Don and Jacobs2010; De Koning et al. Reference De Koning, Gould, Sterk, Tenor, Kunz, Luginbuehl and Seebeck2012; Diaz et al. Reference Diaz, Luengo-Arratta, Seixas, Amata, Devine, Cordon-Obras, Rojas-Barros, Jimenez, Ortega, Crouch, Colmenarejo, Fiandor, Martin, Berlanga, Gonzalez, Manzano, Navarro and Pollastri2014). However, as previously mentioned, field isolates of livestock trypanosome species have proven difficult to establish in culture. Initial cultivation techniques required pre-cultivation over a feeder cell layer of mammalian cells, and subsequently media without the feeder layer were developed (Baltz et al. Reference Baltz, Baltz, Giroud and Crockett1985; Hirumi and Hirumi, Reference Hirumi and Hirumi1994). Recently, metabolic profiling to learn exactly what T. brucei use from their rich culture medium has allowed development of a refined medium for growth of these parasites (Creek et al. Reference Creek, Nijagal, Kim, Rojas, Matthews and Barrett2013). It will be of interest to use the same methods to refine the currently limited media options for T. congolense and T. vivax.
Several in vitro assays have been developed to determine the drug sensitivity of isolates in a faster and cheaper way than in vivo tests (Kaminsky and Brun, Reference Kaminsky and Brun1993). The Alamar blue test (Räz et al. Reference Räz, Iten, Grether-Buhler, Kaminsky and Brun1997) has become the gold standard. Other assays include the drug incubation infectivity test (DIIT) (Kaminsky et al. Reference Kaminsky, Gumm, Zweygarth and Chuma1990) and the [3H]hypoxanthine incorporation test (Brun and Kunz, Reference Brun and Kunz1989), which do not require in vitro adaptation of the trypanosome strain under study, and evaluate parasite viability by their ability to respectively infect mice, or incorporate tritiated hypoxanthine, following 24 h exposure to drug dilutions in a culture medium. Another option for monitoring of resistance onset for T. congolense has been the Drug Incubation Glossina Infectivity Test (DIGIT) (Clausen et al. Reference Clausen, Leendertz, Blankenburg, Tietjen, Mehlitz, Sidibe and Bauer1999): drug resistant and sensitive parasites are distinguished by their ability to infect tsetse flies following their in vitro treatment with specific doses of trypanocidal agents.
Molecular methods
Given the limitations in assessing drug sensitivity levels of veterinary trypanosomes, the development of molecular tests to determine parasites’ susceptibility status would be of profound importance. For T. brucei group parasites it has been shown that mutations in TbAT1 and TbAQP2 genes can underlie resistance to both melaminophenyl arsenicals and to diamidines such as pentamidine (Graf et al. Reference Graf, Baker, Munday, de Koning, Horn and Maser2015; Munday et al. Reference Munday, Settimo and de Koning2015a , Reference Munday, Tagoe, Eze, Krezdorn, Rojas Lopez, Alkhaldi, McDonald, Still, Alzahrani, Settimo and de Koning b ). The TbAT1 gene is also mutated in T. brucei group parasites (including T. b. evansi and T. b. equiperdum) when selected for diminazene resistance (Barrett et al. Reference Barrett, Zhang, Denise, Giroud and Baltz1995; Stewart et al. Reference Stewart, Burchmore, Clucas, Hertz-Fowler, Brooks, Tait, MacLeod, Turner, de Koning, Wong and Barrett2010). Also, in T. brucei loss of the amino acid transporter TbAAT6 underlies resistance to the human African trypanocide eflornithine (Vincent et al. Reference Vincent, Creek, Watson, Kamleh, Woods, Wong, Burchmore and Barrett2010; Schumann et al. Reference Schumann, Jutzi and Roditi2011; Alsford et al. Reference Alsford, Eckert, Baker, Glover, Sanchez-Flores, Leung, Turner, Field, Berriman and Horn2012). PCR-based techniques to assess status of these resistance alleles have, therefore, been possible (Kazibwe et al. Reference Kazibwe, Nerima, de Koning, Maser, Barrett and Matovu2009; Graf et al. Reference Graf, Ludin, Wenzler, Kaiser, Brun, Pyana, Buscher, de Koning, Horn and Maser2013). It has also been possible to develop non-genetic tests for resistance, such as the fluorescence-based test to assess the presence or absence of the P2/TbAT1 transporter (Stewart et al. Reference Stewart, Krishna, Burchmore, Brun, de Koning, Boykin, Tidwell, Hall and Barrett2005).
For T. congolense and T. vivax, however, no reliable markers for drug resistance have yet emerged. As discussed above (see subsection Diminazene aceturate), the assignment of a gene named TcoAT1 as a possible marker for diminazene resistance was erroneous. The MboII–PCR–RFLP was exploited to detect the polymorphism in an ABC-type multidrug transporter putative gene related to isometamidium resistance in T. congolense (Delespaux et al. Reference Delespaux, Geysen, Majiwa and Geerts2005), although this link awaits validation and has not replaced the standard in vivo assays in West Africa (Mamoudou et al. Reference Mamoudou, Delespaux, Chepnda, Hachimou, Andrikaye, Zoli and Geerts2008). However, the possibility that the intrinsic fluorescence of isometamidium could provide a useful marker for resistance to this drug, based on observations that reduced accumulation can underlie resistance, would be useful to follow up; field application would be feasible thanks to the introduction of small but robust, battery-operated fluorescence microscopes, using long-lived light-emitting diodes as fluorescent light sources (Jones et al. Reference Jones, Nyalwidhe, Tetley and Barrett2007).
The paucity of reliable, standardized molecular and diagnostic assays for drug resistance in animal trypanosomes relates to the diversity of infecting species and the difficulties of establishing in vitro cultures for most of them. Neither the basis for drug sensitivity nor drug resistance mechanisms are necessarily shared between the main species that cause AAT (T. brucei spp., T. vivax, T. congolense). Accordingly, it is essential that resistance mechanisms and mode of action models developed in one are properly investigated in the other species, rather than automatically assumed to apply.
NEW COMPOUNDS IN THE PIPELINE
In spite of the economic importance of the veterinary trypanosomiases (in particular of AAT) and of the spreading spectre of drug resistance, new compounds for the diseases have not emerged in many years. A similar difficulty had befallen HAT in the late 20th century, which sparked the emergence of the Drugs for Neglected Diseases initiative (DNDi) and the Consortium for Parasitic Drug Development (CPDD) (Brun et al. Reference Brun, Don, Jacobs, Wang and Barrett2011). These organizations were founded to fill the gap left by the pharmaceutical industry, who judged the investment required to bring new drugs forward to treat diseases of the world's poorest people had no economic rationale. In the case of HAT, the clinical development of pafuramidine, an orally available prodrug of the diamidine furamidine, was halted at an advanced stage due to unforeseen toxicity issues (Paine et al. Reference Paine, Wang, Generaux, Boykin, Wilson, de Koning, Olson, Pohlig, Burri, Brun, Murilla, Thuita, Barrett and Tidwell2010). Since then, DNDi have introduced a more effective combination of two older drugs, eflornithine and nifurtimox (Priotto et al. Reference Priotto, Kasparian, Mutombo, Ngouama, Ghorashian, Arnold, Ghabri, Baudin, Buard, Kazadi-Kyanza, Ilunga, Mutangala, Pohlig, Schmid, Karunakara, Torreele and Kande2009), and brought forward the nitroimidazole fexinidazole (now in phase 2/3 trials as an oral treatment for second stage HAT) and also the benzoxaborole SCYX-7158 (AN5568), another orally available compound with the potential to treat second stage disease (Eperon et al. Reference Eperon, Balasegaram, Potet, Mowbray, Valverde and Chappuis2014). In 2011, the Global Alliance for Livestock Veterinary Medicines (GALVmed), a product development partnership supported by the UK government's Department for International Development (DFID), the Bill & Melinda Gates Foundation and the European Commission, launched a new programme aimed at the discovery of new drugs, vaccines and diagnostics for animal trypanosomiases (https://www.galvmed.org/en/livestock-and-diseases/livestock-diseases/animal-african-trypanosomosis/), on similar principles to the product development partnerships that have aimed to find new treatments for human diseases. A collaboration with Anacor Pharmaceuticals Inc. is hoping to develop and progress a separate benzoxaborole compound to that in human development (Jacobs et al. Reference Jacobs, Nare, Wring, Orr, Chen, Sligar, Jenks, Noe, Bowling, Mercer, Rewerts, Gaukel, Owens, Parham, Randolph, Beaudet, Bacchi, Yarlett, Plattner, Freund, Ding, Akama, Zhang, Brun, Kaiser, Scandale and Don2011; Steinmann et al. Reference Steinmann, Stone, Sutherland, Tanner and Tediosi2015) for the treatment and prevention of AAT. Several other compounds are entering the GALVmed portfolio, as they seek treatments that fulfil important features laid down in a target product profile (TPP), used to assess what properties compounds should have if they are to make an impact in AAT (Table 2).
Table 2. Ideal TPP of a new therapeutic and prophylactic trypanocide for animal African trypanosomiasis [from (http://www.galvmed.org)].
IM, intramuscular; SC, subcutaneous.
Ideally, veterinary drugs, especially in resource-poor and in transhumance societies, should be of high and consistent quality, administrable in single doses sufficient to eliminate or prevent infection, be low cost with high value to the animal's owner, be active on all the relevant species of trypanosomes, have good safety profiles and, for animals destined for human consumption, ideally have very short withdrawal periods. The focus should be on classes of compounds chemically different from existing trypanocides, to minimize potential cross-resistance, and on new compounds selected for slow resistance development.
As efforts to produce new trypanocides and enhanced screening against T. congolense and T. vivax as well as T. brucei spp. are underway, it is becoming increasingly clear that these three species of parasites respond differently to the same compounds, a fact that is likely due to the significant differences in biochemical physiology and membrane transporters of the causative parasites. Different distribution within host tissues will also influence a drug's ability to eradicate an infection. Hence, we can assume that the development of new drugs for the animal trypanosomiases will not be straightforward, will require substantial resources to make progress over a number of years, and will be necessarily linked to increased knowledge of the genetic and phenotypic differences between the three main species of African trypanosomes.
Concluding remarks
Since the introduction of the first veterinary trypanocides more than 60 years ago, treatment of livestock trypanosomiases worldwide has seen barely any innovation, although the available drugs have progressively become less effective and the importance of these infections has not diminished. Indeed, the growing human population and the increasing demand for food (particularly meat and milk) in the tropical and subtropical countries, where these diseases are enzootic, have elevated their importance. Moreover, control of AAT is also becoming an indispensable requisite in the context of the ‘One Health’ approach to eliminate HAT (Simo and Rayaisse, Reference Simo and Rayaisse2015). Hence, research into new curative and chemoprophylactic drugs, with a special focus on efficacy against parasite strains resistant to current treatments, is key.
The search for new veterinary trypanocides would greatly benefit from a better understanding of the biology and the metabolism of animal trypanosomes. This knowledge will be essential to elucidate the mode of action of current trypanocides, understand the molecular factors underpinning resistance, identify new drug targets and quickly screen for new leads. This, in turn, will only be possible if improved laboratory techniques to study these parasites are developed. In particular, the definition of an in vitro culture system for the bloodstream form of T. vivax must be a high priority. At the same time, current empirically formulated culture media used for other trypanosomes should be improved, to better reflect physiological availability of nutrients and make their response to (experimental) drugs more predictive of the in vivo situation, including the rapid detection of resistance development in the field.
While waiting for new trypanocides to become available (which may be some years away) a correct and rational use of the few already licensed drugs is paramount to ensure continuity of their effectiveness. This requires an integrated approach that includes both vector control and appropriate livestock management. Improved, more sensitive diagnostics to promptly detect infected animals, correct treatment of these with the appropriate drug, and improvement of animal general health to help their immunological response to infection, are all important actions to be undertaken that will prolong the useful lifetime of any drug. Use of trypanotolerant breeds (in Africa) and restricting the movement of potentially infected animals (in particular those harbouring mechanically transmitted trypanosomes) are other important control measures to be implemented. The use of sanative pairs (such as diminazene and isometamidium) is essential, although ineffective on its own where multiple-drug resistant trypanosomes are present.
Constant monitoring of drug use and drug resistance appearance will be crucial for correct trypanocide use and to readily improve recommendations for first use and for back up drugs to be utilized in a certain areas when resistance has been confirmed. More efforts and resources will be needed in this field, in order to better understand the extent and the distribution of the trypanosomiases and of resistance to the veterinary trypanocides. The FAO, in collaboration with the International Atomic Energy Agency (IAEA) and the framework of the Programme Against African Trypanosomosis (PAAT) are working in this direction with the launch of the ‘Atlas of tsetse and AAT’, aimed at developing a geospatial database of AAT and Glossina species (Cecchi et al. Reference Cecchi, Paone, Feldmann, Vreysen, Diall and Mattioli2014). For an accurate map of drug resistance occurrence, however, improved tools for its detection in the field are needed, and their development will highly depend on a more thorough understanding of the basic processes that determine resistance onset.
ACKNOWLEDGEMENTS
We are grateful to Fiona Achcar (University of Glasgow) for her assistance with the digital art.
FINANCIAL SUPPORT
F.G., L.J.M., H.D.K. and M.P.B. are funded by the BBSRC (BB/N007999/1 & BB/N007492/1), and also, with T.G.R., by the Global Alliance for Livestock Veterinary Medicines, supported by UKAid (Department for International Development, UK Government) and Bill & Melinda Gates Foundation (UCE-R50A0571 and GAL/0003/0001). FG and MPB are also funded by the Wellcome Trust through a core grant to the Wellcome Trust Centre for Molecular Parasitology (104111/Z/14/Z). LM is a Royal Society University Research Fellow (UF140610) and the Roslin Institute is supported by a core grant from the BBSRC.






 Open access
Open access