



Introduction
The spiral-shaped, Gram-negative microaerophilic bacterium, Helicobacter pylori (H. pylori) colonises the mucosal layer of the stomach epithelium of more than 50% of mankind (Reference Hooi1). H. pylori-human co-existence is ~100 000 years old indicating a great adaptation of H. pylori to its niche in the human gastric mucosa (Reference Moodley2, Reference Camilo, Sugiyama and Touati3). Recently, Hooi et al. and Zamani et al. conducted two separate systematic reviews and meta-analyses on H. pylori's global prevalence (Reference Hooi1, Reference Zamani4). A high prevalence of H. pylori was reported by the first meta-analysis (60.3%); the lowest prevalence was found in Northern America (37.1%) and Oceania (24.4%) and the highest prevalence in Africa (79.1%), Latin America, and the Caribbean (63.4%), and Asia (54.7%) (Reference Hooi1). A lower prevalence was suggested by the second meta-analysis (44.3%), ranging from 34.7% in developed countries to 50.8% in developing countries (Reference Zamani4). The presence of the organism in the oral cavity, water and food introduces them as potential routes of H. pylori transmission (Reference Burucoa and Axon5, Reference Stefano6). Considering that the oral cavity provides an optimal pH, temperature, and microaerophilic environment, oral–oral (particularly mother-to-child) is the prominent mode of H. pylori transmission in both developing and developed countries (Reference Kim and Kim7, Reference Yokota8). H. pylori dwelling in the oral cavity is assumed as a risk factor for the recrudescence of gastric H. pylori infection (Reference Sun and Zhang9). Recrudescence or re-infection leads to recurrence (Reference Sun and Zhang9). Therefore, the recurrence of H. pylori is considered a critical issue (Reference Zhao10). The pathogenicity of H. pylori is attributed to several mechanisms including (i) manipulation of the host signalling pathways, (ii) induction of indirect inflammatory responses within the gastric mucosa, and (iii) induction of direct epigenetic changes on gastric epithelial cells (Reference Machlowska11). Despite inducing an inflammatory response, the human immune system could not eradicate bacterial colonisation, resulting in a life-long infection (Reference Abadi12). H. pylori is a class-I carcinogen and it is known as a main risk factor for intra-gastric diseases including chronic gastritis (CG), peptic ulcer diseases (PUD), gastric cancer (GC), MALT lymphoma (MALToma) and biliary tract cancer (Reference Aviles-Jimenez13-Reference Ota17) as well as extra-gastric diseases, including cardiovascular, metabolic, and neurologic disorders (Reference Franceschi, Covino and Roubaud Baudron18). The complex interplay among the three major factors including: (i) bacterial virulence factors (e.g., cytotoxin-associated gene A (CagA), vacuolating cytotoxin A (VacA) and blood group antigen-binding adhesion (BabA), (ii) host genetic traits (e.g., tumour necrosis factor-alpha (TNF-α), interleukin 10 (IL-10) and IL-1β), and (iii) environmental factors (e.g., high salt, diet, meat consumption and smoke) would result in the diverse clinical outcomes from asymptomatic infection to GC (Reference Chang, Yeh and Sheu19-Reference Toh and Wilson21). Many factors, including geographic regions, living environment, socioeconomic status, educational level, and age are involved in the prevalence of H. pylori (Reference Wang22). Gut microbiome, obesity, male gender, consumption of unpasteurised dairy products, and high-risk occupations (e.g., healthcare and sheepherding) predispose individuals to H. pylori infection (Reference Assaad23-Reference Wang29). The development of various adaptations enables H. pylori to effectively colonise in harsh stomach conditions (Reference Camilo, Sugiyama and Touati3). Using various virulence factors, H. pylori colonises the stomach mucosa, escapes the immune response, and induces the disease (Table 1) (Reference Baj30). H. pylori passes through four steps to colonise, persist and cause serious disease in the human host: (i) overcoming the harsh acidic stomach condition by bacterial urease enzyme, (ii) moving toward epithelium cells by polar flagella and penetrating gastric mucosal barrier, (iii) the interaction of bacterial adhesins (e.g., CagL, CagY, OipA, HopQ, HopZ, BabA/B and SabA/B) with glycan structures (Gly-Rs) exposed on the external surface of gastric epithelial cells and the mucus layer, and (iv) releasing the toxins (e.g., VacA and CagA) causing tissue damages (Reference Kao, Sheu and Wu31). Besides, the virulence factors such as urease, flagellin, arginase and TlpB (a transmembrane chemoreceptor) are survival proteins and the virulence factors including neutrophil-activating protein A (NapA), γ-glutamyl transpeptidase (GGT), peptidoglycan (PG) and ADP-Heptose are immunoresponsive elements (Reference Knorr32). Furthermore, morphological transformation (from spiral to coccoid) is an interesting issue that has been recently considered owing to its vital role in the survival of H. pylori in the host gastric microenvironment (Reference Reshetnyak, Burmistrov and Maev33, Reference Reshetnyak and Reshetnyak34). Although H. pylori numerous virulence factors are responsible for the induction of local damage in gastric mucosa, the host's innate and adaptive immunity including the pattern recognition receptors (PRRs), pro-inflammatory cytokines, chemokines, chemotactic factors, monocytes, macrophages, neutrophils, natural killer cells (NK cells), dendritic cells (DCs), B cells, and T cells facilitate the development of subclinical systemic inflammation (Reference Săsăran, Meliț and Dobru35-Reference Guclu and Agan37). Therefore, the presence of H. pylori in the host gastric mucosa motivates the host's immune system to trigger systemic damage (Reference Mărginean, Mărginean and Meliț38). Despite this strong inflammatory response, H. pylori's ability to evasion, subversion and manipulation of the host immune responses guarantees the development of persistent infection in the stomach mucosa. Alteration in the surface molecules is an efficient strategy that preserves the bacterium from being recognised by the innate immune system (Reference Peek, Fiske and Wilson39). Besides, modulation of the function of the T cells by H. pylori disrupts the host's adaptive immunity against this bacterium (Reference Wen and Moss40). H. pylori prevention and eradication are among the challenging issues in the post-antibiotic era; therefore, providing alternative drugs or vaccine targets is an urgent need (Reference Mohammadzadeh41). In this regard, highlighting the importance of bacterial virulence factors in H. pylori pathogenesis is a promising strategy. This review provides comprehensive data on the H. pylori virulence factors and discusses this bacterium's evasion strategies from the immune response.
Table 1. Virulence factors involving in different aspects of H. pylori pathogenesis (colonization, immune escape, and disease induction), their functions, and associated diseases.
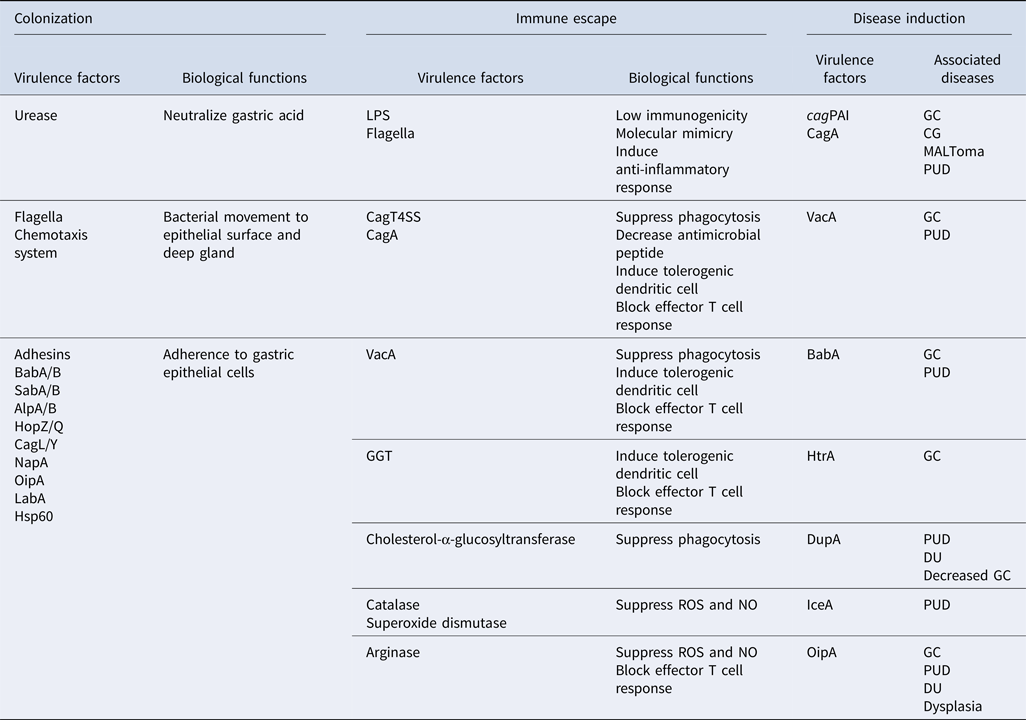
cagPAI, cag pathogenicity island; CagT4SS, Cag type IV secretion system; CagA, cytotoxin-associated gene A; VacA, vacuolating cytotoxin A; HtrA, high temperature requirement A; DupA, duodenal ulcer promoting gene A; IceA, induced by contact with epithelium gene A; OipA, outer inflammatory protein A; GGT, γ-glutamyl transpeptidase; BabA/B, blood group antigen-binding adhesin A/B; SabA/B, sialic acid-binding adhesin A/B; AlpA/B, adherence-associated proteins A/B; HopZ/Q, H. pylori outer membrane protein Z/Q; CagL/Y, cytotoxin associated gene L/Y; NapA, neutrophil-activating protein A; LabA, LacdiNAc-binding adhesin; LPS, lipopolysaccharide; HSP60, heat shock protein 60. ROS, reactive oxygen species; NO, nitric oxide; CG, chronic gastritis; GC, gastric cancer; MALToma, MALT lymphoma; PUD, peptic ulcer diseases; DU, duodenal ulcer.
The genome and virulence factors of H. pylori
The genome of H. pylori was among the first bacterial species which was completely sequenced.
According to the obtained data, H. pylori, strain 26 695, possesses a circular genome of 1 667 867 bp and 1590 predicted coding sequences (Reference Alm42, Reference Tomb JF, Kerlavage, Clayton, Sutton and Fleischmann43). The whole-genome sequencing of H. pylori revealed extraordinary genetic flexibility and a high frequency of gene recombination; this unique nature qualifies the bacteria to survive in harsh and dynamic habitats (Reference Cao44, Reference Noto45). Recognition of virulence factors might pave the way to the illustration of the H. pylori pathogenesis and prediction of the risk for inducing intra- and extra-gastric diseases (Reference Kabamba, Yamaoka and Shiotani46).
Cytotoxin-associated gene pathogenicity island
Cytotoxin-associated gene pathogenicity island (cagPAI) is existent in nearly 70% of all H. pylori strains isolated globally, compared with 60% of western isolates and 95% of East Asian isolates (Reference Olbermann47, Reference Yamaoka48). The cagPAI has been integrated into the H. pylori DNA via horizontal gene transfer (HGT), although its origin is unknown and its genes are not essential for H. pylori (Reference Censini49). cagPAI is a ~40 kb genomic region containing 32 open reading frames (ORFs), namely cag1-26, cagA-Z or cagα-ζ, or by locus name of the HP 26 695 or HP J99 strain genomes (Reference Blomstergren50). cagPAI encode effector protein CagA and type IV secretion system (CagT4SS), a syringe-like structure to inject CagA into gastric epithelial cells (Reference Backert, Tegtmeyer and Fischer51, Reference Chung52). Various bacterial molecules including CagA, DNA and PG metabolites are translocated into host cells by CagT4SS (Reference Backert53-Reference Viala55). Backert et al. suggested that intact CagT4SS consists of a core complex (CagT, CagX, CagM, Cagδ, and CagY), with associated factors (CagH, CagN, CagU, CagV, and CagW); pilus components (CagC, CagH, CagI, CagL, and CagY); and energetic components (CagE, Cagα, and Cagβ). Besides, translocation-associated factors (CagF, CagZ, and Cagβ), and a lytic trans-glycosylase (Cagγ) exist in CagT4SS (Reference Backert, Tegtmeyer and Fischer51). Although the complete composition of cagPAI guarantees encoding of intact CagT4SS, cagPAI is absent in nearly 30% of H. pylori strains, and it is incomplete in some strains (Reference Censini49, Reference Akopyants56). The severity of clinical outcomes induced by H. pylori is dependent on the integrity of cagPAI, therefore partial deletions within cagPAI reduce pathogenic features (Reference Patra57, Reference Nilsson58).
Cytotoxin-associated gene A
According to the presence or the absence of the cagA gene in H. pylori strains two main subpopulations are considered: cagA-positive and cagA-negative strains (Reference Censini49). The co-existence of cytotoxin-associated gene A (CagA)-positive and negative strains in a unique host is not far from expected (Reference Canzian59). Although CagA-positive strains cause more severe clinical outcomes which may progress to malignancy (Reference Baj30, Reference Sharndama and Mba60); the results of a meta-analysis study showed that these strains are more curable (Reference Wang61). CagA oncoprotein is the main virulence factor of H. pylori. Following the adherence of H. pylori to the host gastric epithelial cells, the cagA gene is expressed (Reference Chowdhury62). It was shown that CagA expression is regulated in response to environmental conditions including salt concentration, iron limitation, and pH (Reference Noto63-Reference Loh, Torres and Cover65). Via interaction with different cellular factors, CagA (128–145 kDa) interferes with numerous cellular signal transduction cascades and consequently induces gastric carcinogenesis (Reference Hatakeyama15, Reference Knorr32) (Fig. 1). Molecular anatomy shows that CagA includes a conserved N-terminal region, the variable repeats of the EPIYA (Glu-Pro-Ile-Tyr-Ala) motif, a tyrosine phosphorylation motif, and the C-terminal tail (Reference Hayashi66). According to distinct flanking amino acids around the EPIYA motifs, four different peptide segments, EPIYA-A, B, C and D have been determined (Reference Hatakeyama67). Almost all CagA isolates contain the EPIYA-A and B segments. EPIYA-C segment exists in CagA species that distribute in Europe, North America, and Australia; therefore, it is termed ‘Western CagA’. Also, the EPIYA-D segment exists in CagA species that circulate in East Asia; therefore, it is termed ‘East Asian CagA’ (Reference Hatakeyama68). Upon delivery into the host cell, CagA may attach to the inner surface of the cell membrane and its EPIYA motifs are tyrosine phosphorylated by Src and Abl family kinases, respectively (Reference Backert and Selbach69, Reference Nagase70). Subsequently, it binds Src homology 2 (SH2) domain-containing proteins (e.g., SHP2 tyrosine phosphatase and C-terminal Src kinase (Csk)) and adaptor protein Crk to disturb the adhesion, spreading, and migration of the host cell (Reference Yamazaki71-Reference Higashi74). The induction of NF-κB signalling and IL-8 secretion by CagA and CagT4SS leads to enhanced gastric inflammation, which is considered a risk factor for genetic instability and carcinogenesis (Reference Gorrell75). EPIYA-D motif's more binding tendency to SHP2 compared with the EPIYA-C motif suggests a more intense ability of East Asian CagA for induction of cellular transformations (Reference Hayashi76). Via induction of hypermethylation in the DNA promoters or histones, CagA also mediates epigenetic changes, leading to downregulation of the tumour suppressor genes (e.g., MGMT) or microRNAs (e.g., let-7) (Reference Hayashi77, Reference Sepulveda78). CagA destroys the apoptosis-stimulating protein of the p53 (ASPP2) tumour suppressor pathway and also interacts with the gastric tumour suppressor RUNX3 (RUNX family transcription factor 3), which leads to RUNX3 degradation by the proteasome (Reference Tsang79, Reference Buti80). Distinct CagA species have a different number of CagA-multimerisation (CM) motifs, also called CRPIA (conserved repeats responsible for phosphorylation-independent activity) sites, containing a 16-amino-acid sequence located in the CagA C-terminal region (Reference Ren81). Inside the gastric epithelial cells, the CM motif attaches to and downregulates the polarity-regulating kinase, partitioning-defective 1b (PAR1b), which is also termed the microtubule affinity-regulating kinase 2 (MARK2). The inhibition of PAR1b leads to failure of junction and polarity, which predisposes cells to oncogenesis (Reference Nishikawa82). Carcinoembryonic antigen-related cell adhesion molecules (CEACAMs) are a kind of intercellular adhesion molecules that act as functional receptors for H. pylori outer membrane adhesion HopQ (Reference Javaheri83, Reference Königer84). Recent genetic evidence showed that the interaction between HopQ and CEACAM receptors (CEACAM 1, 5, and 6) is critical for H. pylori CagA translocation while CagT4SS interaction with integrins (neither β1 integrin heterodimers (α1β1, α2β1 or α5β1), nor any other αβ integrin heterodimers) is not essential for this process (Reference Königer84-Reference Zhao86).

Figure 1. Signalling pathways induced by H. pylori CagA. CagA oncoprotein is directly injected into host cells by CagT4SS. This protein interacts with myriad signalling factors to manipulate different signal transduction cascades. CagA, cytotoxin-associated gene A; CagT4SS, Cag type IV secretion system; NFAT, nuclear factor of activated T cells; Bcl-2, B-cell lymphoma 2; c-Met, c-mesenchymal epithelial transition factor; Grb2, growth factor receptor-bound protein 2; GSK3β Wnt, glycogen synthase kinase 3β Wnt; SHP2, Src homology-2 domain-containing protein tyrosine phosphatase-2; TRAF, TNF receptor-associated factor; CD44, cluster of differentiation 44; Akt, Ak strain transforming; BIM, BCL-2-interacting mediator of cell death; MCL1, myeloid cell leukaemia-1.
Vacuolating cytotoxin A
All the H. pylori strains have a single chromosomal copy of the vacA gene. This gene encodes vacuolating cytotoxin A (VacA) protein that forms intracellular vacuoles in the eukaryotic host cells (Reference Foegeding87). vacA gene possesses allelic polymorphism owing to the existence of the signal peptide (s1a, s1b, s1c, and s2 variants), the intermediate (i1, i2, and i3 variants), and the middle regions (m1a, m1b, m1c, and m2 variants) which cause various clinical outcomes and toxic activity in different combinations (Reference Atherton88-Reference van Doorn92). Different combinations of s- and m-regions form four different vacA genotypes including s1m1, s1m2, s2m1, and s2m2; these variants have different abilities to induce vacuolisation in infected cells. The s1m1 variants are the most powerful strains in toxin production and vacuolisation in host cells. Cell vacuolation by s1m2 strains depends on the host cell line, s2m2 strains rarely produce cytotoxin, and s2m1 strains rarely induce cell vacuolation (Reference Pagliaccia91, Reference Rhead93-Reference Atherton96). All s1m1i1 strains are vacuolating and all s2m2i2 are non-vacuolating. S1m2i1 strains induce cell vacuolation, whereas s1m2i2 strains could not do this. Therefore, s1m1i1 and s1m2i1 strains are more virulent and more likely associated with serious clinical outcomes such as GC compared with the s2m2i2 and s1m2i2 strains (Reference Rhead93, Reference Ferreira97, Reference Ferreira, Machado and Figueiredo98). There are extra polymorphic regions including the deletion (d)-region (d1 and d2 variants), located between the i- and the m-region, and the c-region (c1 and c2 variants) located at the 3′-end region of vacA gene (Reference Ogiwara99, Reference Bakhti100). The d1- and c1- genotypes are considered biomarkers of a high risk of GC (Reference Ogiwara99, Reference Bakhti100). It seems that vacA expression is regulated in response to host–microbe interactions, salt and iron concentrations, and low pH (Reference Amilon101-Reference van Amsterdam104). The intact VacA toxin (~88 kDa) consists of an N-terminal domain (33 kDa) and a C-terminal domain (55 kDa) that are involved in cytotoxicity and binding to cell surface receptors, respectively (Reference Torres, McClain and Cover105, Reference Yahiro106). This toxin consists of a signal peptide and it is secreted by the type V secretion system (T5SS, autotransporter) to the intracellular milieu (Reference Chauhan107, Reference Fischer108). Epithelial cell's surface receptors including (i) low-density lipoprotein receptor-related protein-1 (LRP-1), (ii) receptor-like protein tyrosine phosphatase alpha and beta (RPTP-α, -β), and (iii) sphingomyelin acts as receptors for VacA protein (Reference McClain, Beckett and Cover109, Reference Yahiro110). Furthermore, it binds to β2 integrin (CD18) receptors on T cells (Reference McClain, Beckett and Cover109). VacA influences the host cells via induction of apoptosis, autophagy, membrane depolarisation, activation of mitogen-activated protein (MAP) kinases, inhibition of T-cell function and mitochondrial dysfunction contributing to H. pylori life-long colonisation and pathogenesis (Reference Foegeding87, Reference Terebiznik111-Reference Jain, Luo and Blanke116). For triggering apoptosis in the gastric epithelial cells, VacA inserts into mitochondrial membranes, and subsequently, cytochrome C is released (Reference Domańska117). VacA molecule's N-terminally encoded hydrophobic amino acids could form hexameric pores in the lysosomal, endosomal, and mitochondrial membranes of epithelial cells and phagocytes facilitating VacA's pro-apoptotic and vacuolating activity (Reference McClain118). Besides, VacA plays a critical role in H. pylori colonisation via inhibition of the proliferation and activation of B cells and T cells (Reference Torres119). During acute disease, VacA induces the autophagy pathways in host cells, whereas, during chronic disease, it induces the disruption of phagosomes and promotes cell vacuolation that facilitates the survival of H. pylori in the host epithelial cells (Reference Greenfield and Jones120, Reference Raju121).
CagA–VacA interactions
As H. pylori major virulence factors, VacA is expressed by all H. pylori strains, whereas CagA is exclusively expressed by specific strains (Reference Baj30). Deep research in bacterial life suggests a theory that bacteria may release various virulence factors able to act either in a synergistic manner or in an antagonistic manner to achieve greater compatibility with the host (Reference Ricci122). Using a functional relationship between CagA and VacA, H. pylori could fine-tune its interaction with the human host stomach (Reference Ricci122). Most of the time, VacA and CagA cellular activities are antagonistic; CagA downregulates the VacA-induced cellular vacuolation and VacA downregulates the CagA-induced cell alterations (Reference Tegtmeyer123). One of the most important functions of VacA is the induction of autophagy which degrades the CagA protein and shortens its half-life in the host cells. VacA-induced autophagy is downregulated in gastric cells and CagA escapes degradation, such as in the cancer stem cells expressing a cell-surface marker called CD44 variant 9 (CD44v9) (Reference Tsugawa124). Interestingly, a recent report provides contradictory evidence showing that in the absence of VacA, CagA undergoes both proteasomal and autophagic degradation (Reference Abdullah125). VacA causes the accumulation of dysfunctional autophagosomes via disrupting a late step of the autophagic pathway, which results in accumulation (rather than degradation) of CagA in the gastric epithelial cells. It seems that VacA-induced CagA accumulation in dysfunctional autophagosomes results in the vigorous limitation of CagA downstream signalling (Reference Abdullah125). CagA-induced suppression of autophagy via the c-Met-PI3 K/AKT-mTOR signalling pathway enables it to establish pro-inflammatory and carcinogenic action (Reference Li126). CagA obstructs the internalization of VacA to the host cells and leads to the blocking of VacA-induced apoptosis (Reference Akada127). CagA activates calcineurin and subsequently activates the transcription factor nuclear factor of activated T cells (NFAT), which exerts pleiotropic actions on cell proliferation and differentiation. Whereas VacA downregulates the NFAT by decreasing the calcium influx blocking calcineurin activation (Reference Yokoyama128). However, the co-participation of VacA and CagA in the iron acquisition by H. pylori is a rare example of their synergistic effect (Reference Tan129).
Urease
H. pylori urease is involved in the survival and colonisation of the bacterium and induces infection in the stomach (Reference Ansari and Yamaoka130, Reference Voland131). A gene cluster containing seven genes is regulated by two promoters that encode the H. pylori urease enzyme. The ureA and ureB genes (encoding catalytic units) are regulated by the first promoter. Downstream genes, including ureI (encoding acid-gated urea channel) and ure E–H (encoding accessory assembly proteins), are under the control of the second promoter (Reference Akada132, Reference Marcus, Sachs and Scott133). In H. pylori species, urease activity is regulated by the availability of the cofactor nickel and the pH of the stomach microenvironment (Reference Weeks134, Reference Belzer135). It was shown that the existence of nickel in the bacterial culture medium dramatically increases urease activity (Reference Belzer135). Besides, the acid-gated urea channels are closed at pH 7.0 and are firmly open at pH 5.0 (Reference Kao, Sheu and Wu31, Reference Weeks134). Urease is a vital virulence factor to the survival of H. pylori in the human host stomach; recently, Madison et al. demonstrated that in response to nitric oxide (NO), as a product of the host innate immunity, CrdRS two-component system (TCS) regulates the expression of ureA gene (Reference Allen136). Urease is a polymeric enzyme (5–10% of the total protein content) that is composed of two subunits, i.e., UreA (29.5 kDa) and UreB (66 kDa), of which UreB is considered the subunit responsible for the enzyme activity (Reference Strugatsky137). It was shown that a cluster of the 12 active sites containing 24 nickel ions on the urease supramolecular structure guarantees enzymatic action and H. pylori survival at low pH (Reference Ha138). The urease enzyme catalyses the urea conversion to ammonia and carbon dioxide, raising the acidic stomach pH to neutral to protect H. pylori from acidity via the formation of a cloud of ammonia which neutralises acidic pH (Reference Dunn and Phadnis139). Following the production of ammonia, a soluble form of occludin, a 65-kDa tetraspan integral membrane protein, is accumulated which leads to the disruption of tight epithelial junctions (Reference Lytton140). Besides, high levels of ammonia cause cytotoxic effects on the gastric epithelial barrier and ruin these cells’ mitochondrial oxygenation (Reference Schoep141). Natural pH generated by the urease enzyme reduces mucin viscoelasticity to convert the gastric mucin structure from gel to sol, leading to bacterial free movement through the mucus (Reference Celli142). Based on localisation, there are internal and external types of urease enzymes. The internal urease is produced by live bacterial cells and is active at pH 2.5–6.5, whereas the external urease is produced during cell lysis and is active at pH 5.0–8.5 (Reference Scott143). Increased urease activity may lead to a higher risk of induction of histopathological alterations within the gastric mucosa and greater gastric carcinogenesis (Reference Ghalehnoei144).
Helicobacter outer membrane porins
H. pylori adherence to the gastric epithelium is necessary for the delivery of toxins (e.g., CagA and VacA) or other virulence factors into the host cells, which results in inflammatory or immune response-mediated direct or indirect damages (Reference Sharndama and Mba60). The interaction between the receptors expressed on the gastric epithelial cells and Helicobacter outer membrane porins (Hop) family, adhesion factors encoded by hop genes, starts H. pylori infection (Reference Oleastro and Ménard145). Hop family porins including HopS, HopP, HopH, HopQ, and HopZ augment H. pylori adherence to the host cell and boost inflammation by promoting the expression of virulence factors and the secretion of inflammatory cytokines (Reference Xu146). Widely studied among the Hop family are blood group antigen-binding adhesin (BabA) and sialic acid-binding adhesin (SabA) (Reference Doohan147). A wide variety of receptors for BabA and SabA binding activity have been found in the human host stomach and saliva (Reference Xu146, Reference Doohan147). BabA (HopS) binds to H-type 1 and ABO/Lewisb (Leb) blood group antigens exposed on the gastric epithelium and mucus layer (Reference Ansari and Yamaoka148). According to X-ray analysis, it was shown that BabA contains three structural domains including one conserved loop (CL2) and two diversity loops (DL1 and DL2) for interaction with Leb (Reference Moonens149). BabA protein has two domains including the extracellular N-terminal domain linked to Leb antigens and an outer membrane C-terminal domain that anchors into the outer membrane (Reference Hage150). BabA not only contributes to bacterial adherence and colonisation but also augments a nonspecific immune including granulocyte infiltration or secretion of IL-8 enhancing gastric inflammation (Reference Rad151). BabA adherence to fucosylated Leb antigen facilitates CagT4SS activity which leads to the release of high amounts of the pro-inflammatory factors inducing carcinogenesis (Reference Ansari and Yamaoka148, Reference Ishijima152). Low BabA production and decreased binding tendency to the Leb leads to detachment of H. pylori from the gastric mucus and induces ulceration within the duodenum and consequently increases the risk of PUD (Reference Saberi153). The existence of BabA in Western countries is correlated to the high prevalence of PUD and GC, but there is no such correlation in Asians (Reference Chen154). SabA (HopP) is an adhesion molecule, which is involved in H. pylori binding and colonisation via the interaction with sialyl-Lex, sialyl-Lea , and Lex, but not with other Lewis's antigens, such as Lea, Leb or Ley (Reference Mahdavi155, Reference Pang156). SabA-expressing strains could promote gastric diseases, redundant neutrophil infiltration and gastric atrophy during infection; furthermore, these strains could vastly colonise (Reference Sheu157, Reference Yanai158). Recently, it was shown that the tropism of BabA along with SabA for spasmolytic polypeptide-expressing metaplasia (SPEM) glands, enables H. pylori to induce metaplastic alterations and trigger the onset of carcinogenesis (Reference Sáenz, Vargas and Mills159). H. pylori's successful colonisation and persistent infection are largely dependent on two major adhesins: including BabA and SabA. Therefore, these adhesins can be considered potential vaccine candidates against H. pylori (Reference Doohan147, Reference Keikha160-Reference Urrutia-Baca162).
γ-Glutamyl-transpeptidase
γ-Glutamyl-transpeptidase (GGT) is a 61 kDa protein that catalyses the conversion of glutamine into glutamate and ammonia as well as glutathione into glutamate and cysteinyl glycine. This enzyme enables H. pylori to take up and incorporate glutamate into the tricarboxylic acid (TCA) cycle (Reference Ricci163). Initially, it was thought that GGT involves in H. pylori colonisation but more studies confirmed that GGT knockout mutant strains can colonise animals (Reference McGovern164, Reference Oertli165). The GGT expression and activity may promote the development of peptic ulcer disease (PUD) (Reference Gong166). The apoptotic effect on the host cells has been recognised for H. pylori and H. suis GGT (Reference Flahou167). However, it seems that GGT is not the main factor in the induction of H. pylori-mediated apoptosis in T cells because a GGT-negative mutant still induces T-cell apoptosis (Reference Wessler168). H. pylori GGT induces cell cycle arrest at the G1/S phase transition in T cells and gastric epithelial cells, and subsequently inhibits their proliferation (Reference Schmees169, Reference Kim170). It was demonstrated that GGT and VacA could inhibit T cells which emphasises the role of secreted virulence factors in the H. pylori pathogenesis (Reference Wessler168). GGT and VacA promote immune tolerance and gastric persistence that facilitate the prevention of asthma in vivo (Reference Oertli165, Reference Engler171).
Tumour necrosis factor-alpha (TNF-α)-inducing protein
Various H. pylori strains have a conserved sequence of tipα gene encoding tumour necrosis factor-alpha (TNF-α)-inducing protein (Tipα) as a small and secretory protein (Reference Suganuma172). It was shown that Tipα has a weak homology to Gram-positive bacterial penicillin-binding proteins. This led us to conceive that the tipα gene has been derived from Gram-positive bacteria and transferred horizontally to H. pylori (Reference Kuzuhara173, Reference Suganuma174). The Crystal Structure of the Tipα indicates that the functional Tipα protein (approximately 37 kDa) is a homodimer one (Reference Gao175, Reference Jang176). According to the well-established data, a functional T4SS induces epithelial cells to produce cytokines against H. pylori infection (Reference Backert, Tegtmeyer and Selbach177). In addition to the mentioned pathway, it was found that Tipα is a strong inducer of pro-inflammatory cytokine and chemokine gene expressions (Reference Kuzuhara178). Through the activation of nuclear factor kappa B (NF-κB), Tipα induces the overexpression of TNF-α in the Bhas 42 (BALB/3T3 cells transfected with v- H-ras gene) and MGT-40 cells (mouse gastric epithelial cell line) (Reference Suganuma174, Reference Suganuma179). TNF-α is well characterised as a master tumour promotor. It is a key regulator of inflammation that plays a critical role in the cytokine network between cancer and inflammation (Reference Bauer180). Accordingly, Tipα is a potent carcinogenic factor that could induce inflammation and/or CG, hyperplasia, and GC in individuals infected with cagPAI-negative H. pylori strains (Reference Suganuma174, Reference Suganuma179, Reference Morningstar-Wright181). Tipα plays a critical role in the colonisation of mouse gastric mucosa (Reference Godlewska182), however, its secretion is independent of the CagT4SS (Reference Suganuma179). Tipα can bind a membrane receptor called nucleolin and subsequently, it can internalise into the gastric epithelial cell's cytoplasm (Reference Watanabe183). Interestingly, the external ligands of nucleolin including hepatocyte growth factor (HGF), K-ras and Tipα are carcinogenic (Reference Fujiki, Watanabe and Suganuma184). Tipα interacts with single- and double-strand forms of DNA in the GC cells suggesting that DNA binding may have a possible role in the molecular mechanisms of carcinogenesis (Reference Suganuma185). Turning to a monomer form leads to the loss of Tipα's functional performances in tumour promotion, TNF-α induction and NF-kB activation (Reference Suganuma174, Reference Suganuma179). Besides, the monomer form of Tipα has poor DNA-binding ability (Reference Kuzuhara186).
Morphological transformation
H. pylori is typically known as spirally twisted rods, whereas its highly heterogenic nature can form various cell shapes, including coccoid forms, elongated (filamentous) forms, and straight or curved rods (Reference Krzyżek and Gościniak187). During the morphological transition to coccoid forms, H. pylori loses its culturability which could be a sign of bacterial death. Using highly advanced genetic and microbiological techniques, it was shown that these cells are alive and they have modified their physiology (Reference Azevedo188). Decreased cell size and dramatic limitations in metabolic activity lead to H. pylori morphological transformation into coccoid form, which translates into a transition to a viable but non-culturable (VNC) phenotype (Reference Azevedo188). Spiral viable culturable form (SVCF) and coccoid viable but non-culturable form (CVNCF) are two distinct forms distinguished via molecular techniques and electron microscopy (Reference Elhariri189). However, several studies have proposed that coccoid forms colonise the mucus layers, produce virulence factors, escape the immune responses, promote carcinogenesis, and play a critical role in therapeutic failures (Reference Reshetnyak, Burmistrov and Maev33, Reference Sisto190-Reference Chaput195). Therefore, blocking the process of morphological transformation of H. pylori might be a promising strategy for the successful eradication of this pathogen (Reference Krzyżek and Gościniak187, Reference Krzyżek and Grande193). Myricetin (MYR; 3,5,7,3′,4′,5′ hexahydroxyflavone, a natural anti-virulence compound) interferes with the morphological transformation of H. pylori from spiral/rod-shaped forms to coccoid forms and increases the activity of antibiotics against this pathogen (Reference Krzyżek196). It was shown that following exposure to MYR, genes related to the H. pylori morphogenesis (e.g., csd3, sd6, csd4, and amiA) were downregulated. These suppressed genes are mostly involved in the shortening of muropeptide monomers, suggesting their major role in the spiral-to-coccoid transition (Reference Krzyżek196). Cultivation under mild sub-optimal growth conditions such as acidic and alkaline pH, high temperature, aerobiosis, extended incubation, and exposure with a proton pump inhibitor and/or antibiotics stimulates H. pylori morphological conversion from spiral to coccoid (Reference Azevedo188, Reference Shahamat197-Reference Bode, Mauch and Malfertheiner202). It is postulated that commensal bacteria undergo an increased cell filamentation, whereas pathogenic bacteria undergo a reduction of elongation (Reference Rossetti203). In agreement with this hypothesis, it was demonstrated that highly virulent strains had shorter cells than the lower virulent strains; indicating that adaptational changes in cell morphology drastically associate with the virulence profile of H. pylori strains (Reference Krzyżek, Biernat and Gościniak204).
Interaction between H. pylori and the human host immune system: a life-long challenge
Although H. pylori was originally considered an extracellular germ, it was shown that a small portion of this pathogenic bacteria can penetrate the intracellular compartments of various cell types (Reference Ricci, Romano and Boquet205, Reference Dubois and Borén206). It seems that the intracellular lifestyle protects H. pylori from the antibiotics and host immune responses which facilitate persistent infection (Reference Lina207). Despite the induction of intense pro-inflammatory response by various Gram-negative bacteria, H. pylori stimulates only a relatively feeble immune response which results in asymptomatic and persistent infection. Various unique immune evasion strategies mediated by numerous virulence factors are raised H. pylori-derived life-long infection (Reference Sijmons208, Reference Neuper209). These strategies include (i) survival in a harsh habitat, (ii) changing macrophages and DCs functions, (iii) induction of the secretion of the anti-inflammatory cytokines, and (iv) the stimulation of Treg cell development (Reference Kaebisch210-Reference Altobelli213). It was shown that after exposure to live H. pylori, human monocytes initially secret a mixture of pro- and anti-inflammatory cytokines, whereas the prolonged-term innate memory promotes the secretion of anti-inflammatory cytokines (Reference Frauenlob212).
Innate immunity evasion strategies
Escape from innate immune recognition
The highly conserved pathogen-associated molecular patterns (PAMPs) expressed by H. pylori are recognised by PRRs that are expressed by the innate immune cells and gastric epithelial cells (Reference Kawai and Akira214). The interactions between PAMPs and PRRs trigger innate immune responses which are followed by adaptive immune responses (Reference Castaño-Rodríguez, Kaakoush and Mitchell215, Reference Mogensen216). PRR families are classified into transmembrane receptors (Toll-like receptors (TLRs) and C-type lectin receptors (CLRs)) and intracellular receptors (nucleotide-binding oligomerisation domain- (NOD-) like receptors (NLRs) and retinoic acid-inducible gene- (RIG-) I-like receptors (RLRs)) (Reference Akira, Uematsu and Takeuchi217, Reference Takeuchi O218). PAMPs sensation by PRRs activates intracellular signal transduction pathways triggering stormy reactions such as antimicrobial activity and inflammatory response to eliminate the pathogenic microorganism (Reference Akira, Uematsu and Takeuchi217). The TLRs are major classes of PRRs involved in the recognition of PAMPs, such as lipopolysaccharide (LPS), lipoteichoic acid (LTA), hypo-methylated CpG-rich regions of DNA, lipoprotein (LP), flagellin and PG (Reference Takeda and Akira219). Numerous scientific reports explain the role of TLRs during H. pylori infection (Reference Neuper220-Reference Koch222). Since several PAMPs of H. pylori trigger anti- but not pro-inflammatory responses, H. pylori could evade innate and adaptive immune responses to survive and persist in life-long infection (Fig. 2) (Reference Müller and Hartung223). Escherichia coli hexa-acylated LPS with a negative charge and potent immunostimulatory properties activates inflammatory signalling via the sensation of TLR4, whereas H. pylori tetra-acylated LPS with less negative charge could not be recognised by TLR4 and resists antimicrobial peptides (Reference Cullen224). Recently, Schmidinger et al. found that lipid A part of H. pylori LPS interacts with human annexins and dramatically suppresses LPS-mediated TLR4 signal transduction which subsequently hinders the innate immune system (Reference Schmidinger225). The N-terminal position of flagellin in Salmonella enterica could be detected by TLR5, but this critical position (TLR5 binding site) has mutated in H. pylori (Reference Andersen-Nissen226). Additional H. pylori PAMPs-PRRs interactions include the detection of H. pylori hypo-methylated CpG DNA by endosomal TLR9, and sensation of H. pylori LPS by TLR2 (Reference Akira, Uematsu and Takeuchi217, Reference Smith227, Reference Hemmi H, Kawai, Kaisho, Sato, Sanjo, Matsumoto, Hoshino, Wagner, Takeda and Akira228). These two TLRs induce anti-inflammatory and tolerogenic responses causing persistent infection (Reference Otani229-Reference Sun231). H. pylori 5ʹ triphosphorylated RNA is recognised by endosomal TLR8; this triggers a downstream signalling pathway that results in the transactivation of type I interferon (IFN-I (IFN-α/β)) in human monocytes (Reference Akira, Uematsu and Takeuchi217, Reference Lee232). H. pylori 5ʹ triphosphorylated RNA is also detected by one kind of RLRs known as RIG-I. The stimulation of the RIG-I receptor leads to the activation of the transcription factors IRF3 and IRF7 and subsequent expression of IFN-I (Reference Rad233). According to in vitro and ex vivo experiments, recently it was shown that through the downregulation of IRF3 activation, H. pylori actively inhibits cyclic GMP-AMP (cGAMP) synthase (cGAS)-stimulator of interferon genes (STING) and RIG-I signalling (Reference Dooyema234). DCs-specific intercellular adhesion molecule-3 grabbing non-integrin (DC-SIGN) is a member of the CLR family which plays a critical role in H. pylori recognition and pathogenesis. DC-SIGN ligands are mannosylated in most of the pathogens causing activation of the pro-inflammatory pathways. Although DC-SIGN ligands of H. pylori are fucosylated leading to the suppression of the signalling pathways downstream of DC-SIGN and also causing activation of the anti-inflammatory genes (Reference Gringhuis235). The macrophage-inducible C-type lectin (MINCLE) is another CLR, it interacts with the Lewis antigens of H. pylori LPS inducing macrophages to secrete low levels of TNF-α and high levels of IL-10 promoting H. pylori persistence (Reference Devi, Rajakumara and Ahmed236). NOD1 (CARD4) and NOD2 (CARD15) are two members of NLRs that can recognise various motifs in PG of various Gram-negative pathogenic bacteria (Reference Girardin237, Reference Girardin238). PG delivered by the H. pylori CagT4SS is sensed by NOD1 in the cytoplasm of epithelial cells and causes the activation of NF-κB signalling and upregulation of pro-inflammatory immune responses (Reference Viala55). It was shown that the immunomodulatory glycoprotein olfactomedin 4 (OLFM4) targets NOD1 and NOD2 in H. pylori-infected cells (Reference Liu239). OLFM4 is a target gene of the NF-κB pathway and associates directly with both NOD1 and NOD2 proteins thereby having a negative feedback effect on NF-κB activation induced by H. pylori infection. It was shown that the knockout of the OLFM4 gene in mice reduces H. pylori loads and increases gastric immune cell infiltration. Via a negative regulatory effect on H. pylori-specific NOD-mediated immune responses, OLFM4 plays an impressive role in the persistence of H. pylori colonisation (Reference Liu239). The bacterial ligand such as PG can access intracellular NOD-1 by CagT4SS and outer membrane vesicles (OMVs) (Reference Minaga240). PG deacetylation allows H. pylori to evade host innate immunity via induction of an NOD-1-dependent negative feedback loop. It seems that NOD-1 alters macrophage polarisation leading to H. pylori persistence (Reference Suarez241). Bacterial sugars including ADP-β-D-manno-heptose (ADP-Heptose) and D-glycero-β-D-manno-heptose-1,7-bisphosphate (βHBP) are key intermediate metabolites of LPS inner heptose core. The biosynthesis of ADP-Heptose and βHBP is a novel potent and cagPAI-mediated cell activation pathway triggering NF-κB augmentation and IL-8 release in human epithelial cells (Reference Pfannkuch242, Reference Zimmermann243). Upon CagT4SS-mediated internalization of βHBP into the gastric epithelial cells, it is sensed by α-kinase 1 (ALPK1) and TNF receptor-associated factors (TRAF)-interacting protein with forkhead-associated domain (TIFA). The activation of the ALPK1-TIFA signalling axis leads to the activation of NF-kB signalling (Reference Pfannkuch242-Reference Zhou244). Recently, Maubach et al. showed that the TIFA has dual functions in H. pylori-induced NF-κB pathways; induction of NF-κB classical pathway via the association of TIFA with TRAF6, and induction of NF-κB alternative pathway via the association of TIFA with TRAF2/TRAF3 (Reference Maubach245). The TIFA is an intrinsic anti-parallel dimer, containing a threonine residue (Thr9), a central forkhead domain (FHA), and a C-terminal TRAF6 binding site (T6BP). Following βHBP sensation, Thr9 is phosphorylated and then intermolecular pThr9–FHA interactions lead to head-to-tail oligomerisation. The TIFAsome (including TIFA oligomers, TRAF2, and additional host factors) recruits and activates TRAF6, triggering NF-κB activation and subsequent inflammatory signalling (Reference Gaudet and Gray-Owen246). Following TIFA activation, PG delivered through the CagT4SS activates NOD1 which leads to NF-κB-mediated pro-inflammatory responses. Within hours of infection and before NOD1 activation, the ALPK1-TIFA signalling pathway is activated and triggers strong NF-ҡB-dependent inflammation. Finally, the interaction of CagA (delivered through the CagT4SS) and host transforming growth factor (TGF)-β-activated kinase 1 (TAK1) triggers NF-κB-mediated pro-inflammatory responses (Reference Gall247). Fig. 3 illustrates the model of H. pylori CagT4SS-mediated, NF-κB-driven innate immune response in gastric epithelial cells. The successive activation of the ALPK1-TIFA signalling pathway and NOD1, and CagA delivery trigger the initial inflammatory response in gastric epithelial cells, motivate the subsequent recruitment of immune cells and lead to CG (Reference Gall247); however, the contribution rates of these pathways to natural infection are not clear. It was reported that mutation in the genes required for the synthesis of βHBP (e.g., rfaE, gene HP0858 in strain 26 695) dramatically reduces IL-8 induction (>95%) and ruins CagT4SS-dependent cellular signalling (Reference Tomb JF, Kerlavage, Clayton, Sutton and Fleischmann43, Reference Stein248). It was shown that ADP-Heptose, a derivative of βHBP, was present in H. pylori at more concentration (10 times) than its origin. Simultaneously, ADP-Heptose was dramatically more potent and cells distinctly recognise the existence of the β-form. The aforementioned findings revealed that ADP-Heptose is not only a new potent NF-kB-activating PAMP in H. pylori but also it is a general Gram-negative bacteria-derived PAMP (Reference Pfannkuch242). Furthermore, ALPK1 is a cytosolic innate immune receptor for bacterial ADP-Heptose (Reference Zhou244). The activation of the ALPK1-TIFA-NF-kB axis and CagA translocation are two distinct functions of CagT4SS because, despite the mutation in bacterial rfaE or the host ALPK1, CagA internalization continues (Reference Zimmermann243). It was demonstrated that cagPAI proteins enhance the colonisation rate via the suppression of antimicrobial peptides (Reference Patel249). The inverse correlation between gastric β-defensin 1 level and colonisation in H. pylori carriers is attributed to the reduced expression of human β-defensin 1 in a CagT4SS-dependent manner (Reference Patel249). β-defensin 3 is another human antimicrobial peptide, with strong anti-H. pylori activity. At the beginning of in vitro infection, β-defensin 3 is induced in an MAP kinase- and epidermal growth factor receptor (EGFR)-dependent manner. The induction of β-defensin 3 is followed by the stable shutdown through CagA-mediated activation of the Src homology domain, containing protein tyrosine phosphatase 2 (SHP2) and downmodulation of the EGFR signalling pathway (Reference Bauer250).

Figure 2. H. pylori evasion of innate immune recognition. Structurally modified pathogen-associated molecular patterns (PAMPs) enable H. pylori to evade the detection by pro-inflammatory Toll-like receptors (TLRs). H. pylori tetra-acylated LPS is less biologically active and is not sensed by TLR4. The TLR5 cannot detect the mutated TLR5 binding site of H. pylori flagellin. TLRs 9 detects H. pylori DNA (CpG DNA) and TLR2 detects H. pylori LPS; these TLRs predominantly activate anti-inflammatory signalling pathways and IL-10 expression. The cytosolic receptor RIG-I and endosomal receptor TLR8 sense 5ʹ triphosphorylated RNA which elicit IFN-α/β response. H. pylori fucosylated DC-SIGN ligands are another activator of anti-inflammatory genes. Besides, theses ligands block IFN-α/β response. Notice that the different cell types express various pattern recognition receptors (PRRs). TLR2/4/5/8/9, Toll-like receptor 2/4/5/8/9; LPS, lipopolysaccharide; DC-SIGN, dendritic cell-specific intercellular adhesion molecule-3 grabbing non-integrin; SRC, steroid receptor coactivator; IFN-α/β, α/β interferon; IRF3/7, interferon regulatory factor3/7; AP-1, activator protein 1; NF-κB, nuclear factor-κB; P50/P65, NF-κB p50/p65 heterodimer; TIR, Toll/interleukin-1 receptor domain; MYD88, myeloid differentiation primary response gene 88; DD, death domain; CARD, caspase activation and recruitment domain; RIG-I, retinoic acid-inducible gene I, CpG DNA, 5'—C—phosphate—G—3'.
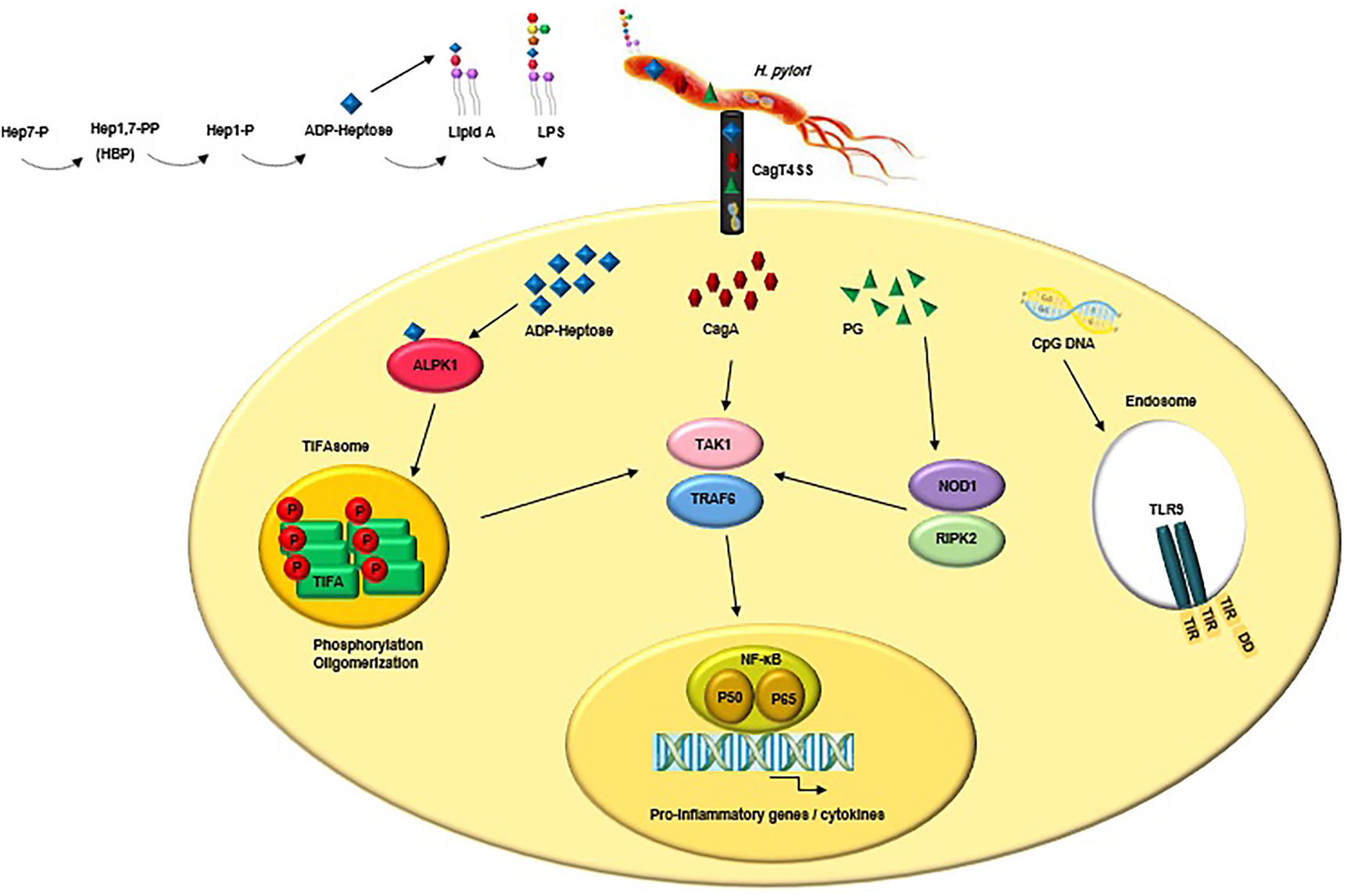
Figure 3. CagT4SS-injected virulence factors (e.g., ADP-Heptose, CagA and PG) trigger NF-κB-mediated innate immune response in gastric epithelial cells. Upon binding of ADP-Heptose, an intermediate metabolite produced during the biosynthesis of LPS, to ALPK1, the ALPK1-TIFA signalling pathway is triggered, which results in an NF-kB-dependent pro-inflammatory response. Following TIFA activation, PG activates NOD1, which leads to NF-κB-mediated pro-inflammatory responses. Finally, the interaction of CagA and host TAK1 triggers NF-κB-mediated pro-inflammatory responses. Besides, CagT4SS-injected CpG DNA is detected by TLR9 which result in activation of anti-inflammatory signalling pathways. LPS, lipopolysaccharide; CagT4SS, Cag type IV secretion system; ADP-Heptose, ADP-β-D-manno-heptose; PG, peptidoglycan; CagA, cytotoxin-associated gene A; ALPK1, alpha kinase 1; Tak1, TGF-β activated kinase 1; TRAF6, tumour necrosis factor receptor (TNFR)-associated factor 6; TIFA, TRAF interacting forkhead-associated protein A; NOD1, nucleotide-binding oligomerisation domain 1; RIPK2, receptor-interacting-serine/threonine-protein kinase 2; NF-κB, nuclear factor-κB; P50/P65, NF-κB p50/p65 heterodimer; CpG DNA, 5'—C—phosphate—G—3'; TLR9, Toll-like receptor 9; TIR, Toll/interleukin-1 receptor domain; DD, death domain.
Manipulation of innate immune cells
The innate immune cells in the lamina propria and the mucus layer on the surface of gastric epithelial cells are the main defensive barrier against H. pylori (Reference Chmiela251). H. pylori CagA in macrophages interacts with SHP-1 and targets TRAF6 for K63-linked ubiquitination, thereby this multi-functional protein can down-regulate the expression of pro-inflammatory cytokines and subsequent immune response (Reference He252). Gang Liu et al. showed that cagA-positive H. pylori strains abrogate Cathepsin C (CtsC) to ruin neutrophil activation; this process prevents bacterial clearance and guarantees persistent infection (Reference Gang Liu253). It was shown that the phosphorylated form of CagA over-expresses the gene encoding haem oxygenase-1 which leads to macrophage anti-inflammatory responses and causes persistent infection (Reference Gobert254). It was demonstrated that CEACAM1 expressed mainly by activated immune cells, including NK cells and T cells acts as a suppressive receptor (Reference Gray-Owen and Blumberg255). Recently, Gur et al. showed that the secretion of IFN-γ by CD4+ T cells is suppressed by HopQ; also, NK cell and T-cell functions are inhibited by HopQ-mediated activation of CEACAM1 which may result in immune cells’ harness (Reference Gur256). HopQ–CEACAM interaction modulates the expression and secretion of chemokines in immune cells which leads to H. pylori's survival within neutrophils in a HopQ-dependent manner (Reference Behrens85). Via the suppressive effects on DCs and inductive effect on macrophages, VacA suppresses IL-23 expression and induces IL-10 and TGF-β secretion. The VacA immunomodulatory activity built a tolerogenic environment for H. pylori and leads to a chronic infection (Reference Altobelli213). The reduction of proliferation of immune cells, including neutrophils, macrophages, eosinophils, DCs, B cells, and T cells is also attributed to the VacA protein (Reference Foegeding87, Reference Djekic and Müller257, Reference Utsch and Haas258). VacA downregulates class II major histocompatibility complex (MHC II)-dependent pathways and via the formation of vesicular compartments inside macrophages assists H. pylori's intracellular survival (Reference Zheng and Jones259). NapA manipulates both innate and adaptive immune responses by: (i) upregulation of MHC II, (ii) promotion of T helper1 (Th1) cell differentiation, and (iii) stimulation of IL-12 and IL-23 release from neutrophils and monocytes (Reference Amedei260). Another potent immunomodulator is the urease enzyme; it (i) modifies opsonisation, (ii) augments the chemotaxis of neutrophils and monocytes, (iii) binds to the MHC II receptors, and subsequently facilitates apoptosis, and (iv) increases the secretion of the pro-inflammatory cytokines (Reference Schmalstig261). GGT downregulates T-cell proliferation and DCs differentiation (Reference Oertli165, Reference Schmees169, Reference Gerhard262, Reference Beigier-Bompadre263). To achieve life-long survival in the host, H. pylori downregulates macrophage phagocytosis and inhibit NO production by the induction of macrophage arginase II (ARG2) (Reference Ramarao264, Reference Lewis265). Inhibition of cell division-associated genes by H. pylori attenuates the proliferation of macrophages (Reference Tan266). Besides, H. pylori promotes mitochondrial membrane depolarisation and hydrogen peroxide secretion which induce apoptotic program in macrophages (Reference Chaturvedi267, Reference Asim268). Persisting a life-long H. pylori infection in the host depends on the inhibition of macrophage-mediated functions including phagocytosis, human leukocyte antigen-II (HLA-II) expression and IFN-γ production which results in T-cell suppression (Reference Cheok269). Via the glucosylation of cholesterol, H. pylori escapes from macrophage phagocytosis and could survive (Reference Yang and Hu270). Morey et al. showed that H. pylori depletes cholesterol in gastric glands to inhibit IFN-γ signal transduction and evade inflammatory response (Reference Morey271). Antigen presentation to T cells is one of the most important functions of DCs; abrogation of this function by H. pylori blocks Th1 cell differentiation (Reference Mitchell272).
Adaptive immunity evasion strategies
Despite the high prevalence of H. pylori infection among the human race, the majority of the infected individuals are asymptomatic for life, and only a minority of them develop H. pylori infection-related disease (Reference Müller and Hartung223, Reference Alexander273). Studies on human carriers and mouse models show that the polarisation and severity of the H. pylori-specific Th cell responses are major predictors and drivers of the disease (Reference Arnold274). Compared with the asymptomatic carriers, the PUD patients have a threefold higher anti-H. pylori Th1 cell response, twofold lower Treg response, sixfold higher Th2 response, and dramatically reduced levels of TGF-β and IL-10 in the gastric mucosa (Reference Robinson275). This imbalance suggests an association between inadequate Treg response and the development of H. pylori-derived disease (Reference Robinson275). It was shown that the severity of gastritis has an inverse correlation with the number of gastric Tregs and the cytokines secretion by Tregs (Reference Harris276). These findings are in line with previous work showing that Tregs gather in the gastric mucosa and quench exclusive memory T-cell responses in the infected but not uninfected patients (Reference Lundgren277, Reference Lundgren278). Treg responses to the infection are predominantly launched in the asymptomatic (healthy) carriers resulting in effectively damping immunopathologic reactions and promotion of the persistent infection. Although T-effector-dominated responses are predominantly expressed in symptomatic carriers that promote the disease (Reference Müller and Hartung223). Using two critical cytokines including IL-10 and TGF-β, Treg cells can inhibit T-effector cell-driven immunopathology (Reference Arnold274). Thus, the expression level of these cytokines in the gastric mucosa could effectively predict H. pylori-induced clinical outcomes (Reference Müller and Hartung223). It was shown that the downregulation or neutralisation of IL-10 signalling could effectively trigger strong T-cell-dependent immunopathology and clear H. pylori (Reference Sayi279). Comparison of T-cell responses of the symptomatic versus asymptomatic carriers as well as children with mild gastritis versus adults with severe gastritis suggests that Treg/T-effector cell ratios are associated with the clinical outcome (Reference Robinson275, Reference Harris276). Since vaccination is the only effective strategy to achieve protective immunity, understanding the importance of T-effector versus Treg responses to establish H. pylori clearance or immunopathology is a major issue in H. pylori vaccinology (Reference Hitzler280). For successful colonisation, H. pylori must suppress effector T cells (Th1 and Th17 subsets) activity, proliferation and clonal expansion. GGT and VacA are two critical virulence factors devastating T-cell-mediated immunity (Reference Müller and Hartung223) (Fig. 4). β2 integrin subunit of the heterodimeric transmembrane receptor lymphocyte function-associated antigen-1(LFA-1) acts as a receptor for hexameric VacA (Reference Sewald281). Subsequent ligand–receptor binding, VacA is entered upon protein kinase C-mediated serine/threonine phosphorylation of the β2 integrin cytoplasmic tail (Reference Sewald, Jiménez-Soto and Haas282). Cytoplasmic VacA blocks NFAT dephosphorylation by the Ca2+/calmodulin-dependent phosphatase calcineurin. Then the nuclear transfer of NFAT was prevented resulting in the downregulation of IL-2 production as well as subsequent T-cell activation and proliferation (Reference Müller and Hartung223). GGT could arrest T-cell proliferation in the G1 phase of the cell cycle through the disruption of the Ras signalling pathway (Reference Lina207). Using a cAMP-dependent pathway, VacA and GGT trigger the secretion of miR-155 and Foxp3 in the human lymphocytes (Reference Fehri283). The GGT enzymatic activity is linked to its immunomodulatory effects, whereas VacA vacuolating cytotoxicity is independent of its immunomodulatory effects, as both the non-toxigenic (s2/m2) or toxigenic (s1/m1) types of VacA are equally tolerogenic in vitro (Reference Oertli165). Selective recruitment or activation of Tregs in the preferred niche facilitates the promotion of chronicity by persistent pathogens, such as Mycobacterium tuberculosis and certain helminths (Reference McBride, Konowich and Salgame284). The same is true for H. pylori, which establishes Treg-mediated immunosuppression promoting chronic infection (Reference Arnold274, Reference Robinson275). In vitro and in vivo findings imply that H. pylori not only triggers DCs-derived tolerogenic (i.e., Treg-inducing) responses but also quenches their immunogenic functions (Reference Müller and Hartung223). Mature and immature DCs diversely affect the Th cells differentiation which results in immunity or immunosuppression (Fig. 5). DCs maturation is promoted via the cytosolic or membrane-bound PRRs-mediated recognition of PAMPs. Mature DCs can express high levels of MHC II, maturation markers, co-stimulatory markers (e.g., CD80, CD86, and CD40), Th cell-activating cytokines, and Th cell-differentiating cytokines as well as other pro-inflammatory cytokines (e.g., IL-12, IL-23, TNF-α, and IL-6) (Reference Kaebisch210). Differentiation of naive T cells into Th1 or Th17 cells depends on antigen recognition via the T-cell receptor, soluble cytokine signals, co-stimulatory signals, and high-level of IL-12 and IL-23 (Reference Müller and Hartung223). This scenario will result in immunity and H. pylori control. H. pylori-exposed DCs remain immature and present tolerogenic activity. Despite the high expression of MHC II by the semi-mature DCs, these cells fail to express co-stimulatory markers and Th1/Th17 cells differentiating cytokines, and instead, they produce high levels of the anti-inflammatory cytokine IL-10 (Reference Oertli285). These semi-mature DCs efficiently induce Treg differentiation and immunosuppression in vitro and in vivo (Reference Oertli285); however, LPS treatment can break the tolerance in vivo (Reference Oertli285). The H. pylori virulence and persistence factors including VacA and GGT act as the inhibitors of murine DCs maturation and tolerogenic reprogramming (Reference Oertli285); CagT4SS is their counterpart, which acts on human DCs (Reference Kaebisch210). Mutation of either GGT or VacA (i) reduces the colonisation of mutant strains in mice, (ii) suppresses the prevention of LPS-induced DCs maturation, and (iii) inhibits the DCs tolerisation (Reference Oertli165). Via a partial or total inhibition of activation of T cells in the lamina propria, VacA proteins can subvert the immune response (Reference Reshetnyak, Burmistrov and Maev33, Reference Raju121). All in vivo findings suggest that under H. pylori forces, DCs present tolerogenic properties leading to the anti-inflammatory cytokine secretion and Treg differentiation, as well as downregulation of T-effector cell function, and establishment of persistent infection (Reference Müller and Hartung223). Tregs facilitate the persistence of infection and protect the infected host cells against gastric inflammation. Besides, Tregs encourage bacterial colonisation which likely promotes gastric tumour progression (Reference Laur286).

Figure 4. H. pylori subverts T-cell-mediated immunity using the secreted virulence factors VacA and GGT. Following the internalization, Cytoplasmic VacA prevents nuclear translocation of NFAT by inhibiting its dephosphorylation by the Ca2+/calmodulin-dependent phosphatase calcineurin and thereby blocks IL-2 production and subsequent T-cell activation and proliferation. The GGT prevents the proliferation of T cells via interfering in the G1 phase of the cell cycle. TCR, T-cell receptor; MHCII, major histocompatibility complex class II; VacA, vacuolating cytotoxin A; GGT, γ-glutamyl transpeptidase; NFAT, nuclear factor of activated T cells; IL-2, Interleukin-2; CnA/B, calcineurin A/B subunits; CaM, calmodulin; LFA-1, lymphocyte function-associated antigen-1; PKCζ/ η, protein kinase Cζ/η.

Figure 5. The immunological response elicited by H. pylori infection. H. pylori is genetically highly variable and expresses various virulence factors, including adhesins (BabA/SabA), enzymes (urease), toxins (VacA) and effector proteins (CagA) which are involved in bacterial pathogenesis. The immunity and immunosuppression are the consequences of the maturation status of dendritic cells (DCs), which direct T helper cell differentiation. CagT4SS, Cag type IV secretion system; CagA, cytotoxin-associated gene A; VacA, vacuolating cytotoxin A; BabA, blood group antigen-binding adhesin A; SabA, sialic acid-binding adhesin A; DC, dendritic cell; Mφ, macrophage cell; PMN, polymorphonuclear leukocyte; Treg, regulatory T cell; Th1/17, T helper cell1/17; GM-CSF, granulocyte–macrophage colony-stimulating factor; TNF-α, tumour necrosis factor-α; IFN-γ, interferon-γ; TGF-β, transforming growth factor-beta, CXCL1/2, C-X-C motif chemokine ligand 1/2; IL-1/2/6/8/10/12/17/23, interleukin 1/2/6/8/10/12/17/23.
Vaccine development against H. pylori
Owing to high antibiotic resistance among H. pylori strains, World Health Organization (WHO) put this bacterium on its high-priority pathogen list to prioritise research and development on the deadly threats to global health (Reference Tacconelli287). Owing to increasing antibiotic resistance, high cost, poor patient compliance, and re-infection the current therapeutic strategies remain sub-optimal (Reference Megraud288). Besides, GC is the most drastic clinical outcome of H. pylori infection, and it is ranking fifth for incidence and fourth for mortality globally (Reference Sung289). Therefore, priority for H. pylori eradication comes with vaccination (Reference Hooi1, Reference Mohammadzadeh41, Reference Yoon290). Until now various kinds of vaccines including (i) inactivated whole-cell vaccines, (ii) subunit and recombinant protein vaccines, (iii) epitope-based vaccines, (iv) DNA vaccines, and (v) vector vaccines have been introduced for vaccination against H. pylori (Reference Soudi291-Reference Ansari, Tahmasebi-Birgani and Bijanzadeh295) (Fig. 6). Reviewing the recent progress on H. pylori vaccines shows that these efforts have not been very fruitful and most vaccine candidates are at a very early stage (phase I or even preclinical) (Reference Mohammadzadeh41, Reference Zhang296, Reference Dos Santos Viana297). H. pylori evasion from the host adaptive immunity and its persistent infection are attributed to extreme adaptation to the gastric environment and result in current vaccines’ partial or limited protectivity (Reference Camilo, Sugiyama and Touati3, Reference Robinson, Kaneko and Andersen298). There is no agreement on the role of humoral immunity in H. pylori vaccine-induced protective immunity (Reference Guo299, Reference Akhiani300). Nevertheless, Sun et al. showed that Th1 and Th17 cell responses induced by immunodominant antigens protect mice against H. pylori infection which confirms the vital role of cellular immunity for the clearance of H. pylori infection (Reference Sun301). Acquisition of H. pylori infection occurs in early childhood, so an effective vaccine should protect individuals for 10–15 years and more (Reference Sutton302). H. pylori's successful colonisation, persistence and induction of severe clinical outcomes in the human host is dependent on passing through several steps (Reference Kao, Sheu and Wu31). Designing and developing multivalent and multistage vaccines containing various immunodominant antigens involved in different aspects of H. pylori colonisation and pathogenesis could be a hopeful strategy for the eradication of this insidious bacterium (Reference Mohammadzadeh41).

Figure 6. Vaccine development against H. pylori. CagA, cytotoxin-associated gene A; VacA, vacuolating cytotoxin A; BabA, blood group antigen-binding adhesin A; HspA, heat shock protein A; FliD, Flagellar hook-associated protein 2.
Conclusions
H. pylori is an uninvited guest in the stomach mucosa of more than 50% of mankind. Variable morphology and expression of numerous heterogenic virulence factors are two intrinsic traits that make H. pylori a successful pathogen in the hostile stomach environment. Using various mechanisms, H. pylori manages to escape from the innate and adaptive immune responses. In this condition, the immune response cannot eradicate the bacterium and facilitates bacterial colonisation and survival in the gastric mucosa (Reference Abadi12, Reference Lina207, Reference Mejías-Luque, Gerhard and Tegtmeyer303). Therefore, immune evasion makes vaccination a challenging issue. In the majority of infected individuals, H. pylori and its host are in balance leading to chronic infection without any serious clinical outcomes. However, this balance is not universal, and individuals develop various clinical outcomes from asymptomatic infection to GC. The interaction between the environmental factors (e.g., diet and smoke), host genetic polymorphism (e.g., IL-1β, IL-10, and TNF-α), and the H. pylori virulence factors (e.g., CagA and VacA) will determine the clinical outcomes in individuals. So far, a wide range of virulence factors have been attributed to H. pylori classified as adhesins (BabA), enzymes (urease), toxins (VacA), and effector proteins (CagA) to influence the host-pathogen interactions. The induction of chronic persistent infection despite a strong host immune response is a unique feature of H. pylori pathogenesis. The identification of the host inflammatory pathways that H. pylori activates and subverts might help to understand the complicated pathogenesis of H. pylori. Therefore, the profound understanding of the exclusive role of H. pylori virulence factors in (i) induction of various clinical outcomes and (ii) H. pylori–host interaction to subvert the immune response will pave the way for alternative therapies and vaccine development.
Author contributions
All authors contributed to the writing and review of the manuscript.
Disclosure statement
The authors report no conflicts of interest.
Funding
This work was supported by the National Institute for Medical Research Development (NIMAD) (Grant no. 989320).







