



Recommendations for a New Core Facility Manager
Confocal Listserver
Dear All, I recently secured a new role as an imaging facility manager, and I will start in the coming month. I am looking for recommendations from anyone that works in imaging facilities. What are the vital lessons you learned that you wished you knew when you started in the facility? Similarly, what courses, books, or resources would you recommend to new managers like me? I look forward to hearing from everyone. Kind regards, Alicerita Eseola [email protected]
I have 2 suggestions: 1) I suggest you not to charge any fee for training new users. This has several benefits. More group leaders will send their users to the facility to learn microscopy, and your facility will blossom. A successful core facility is a busy core facility! Having many users from various research groups will secure a higher rank of importance for your facility within your organization and this will have several benefits. For example, finding financial support for a broken and expensive piece of equipment will not only be your problem. You will have priority when there are funds or space to be shared. The groups using your facility will also help you by lobbying or by direct support when you need help. The most important benefit is that nobody will shy away when needing basic or advanced training from you, hence all users will be trained properly by a professional rather than trying to learn from other members in their group or by themselves. Certainly, never allow anybody to use instruments without at least basic training (paid or not). However, providing “knowledge” for free will make you more approachable. This will immensely reduce mishaps and mistreatment of equipment. 2) With regard to mistreatment of equipment, there should always be facility personnel available to provide in-person help or via a phone call in case of trouble. Imaging facility instruments are complex. During late hours or weekends, users should not be afraid to call facility staff. Ferhan Ayaydin [email protected]
We from German BioImaging are organizing a Core Facility Leadership and Management Course once per year in Düsseldorf, Germany, together with trainers from hfp consulting. The next course is planned for June 2023. During the course, which is specifically tailored for core facility staff and managers, participants will learn to improve their leadership and management skills. For details, feel free to visit our webpage: https://gerbi-gmb.de/activities/core-facility-leadership-and-management-course/ or the course website from this year: https://gerbi-gmb.de/event/gerbi-gmb-core-facility-leadership-and-management-course/. The course has now run for almost 10 years and has received excellent feedback from the participants. Feel free to reach out if you have any questions (contact details are below). I wish you all the best for your future journey! Janina Hanne [email protected]
What I wished I had known when I started our facility from scratch, 15 years ago:
- Do not buy all equipment at once (I did not do that, and I am really happy about it). When one starts at a facility, one might not be fully aware of the actual needs of the pool of users. Acquiring this ‘local’ knowledge takes time. It is a real pity to buy equipment that is not really needed and therefore is seldom used.
- Make a real market survey before you decide to buy any new system and demand a thorough demo, if possible, in house and for 2 weeks so your users get to test with their samples.
- Spend a lot of time with users when a new instrument arrives. This in my opinion is a great way for the facility staff to get fully accustomed to new systems and gain experience with all sorts of samples and requirements. This is also important to discover bugs and problems early so one gets a chance to complain and get them fixed.
- Devote proportionally more time and energy to the most edgy projects compared to typical ones. Edgy projects might represent only 2% of the usage of the facility but they are what keeps you interested, what makes you an expert, and finally what drives the facility upwards. All users will benefit from these 3 points.
- We believe that it is important for users to pay for their training. To me there is not a reason to provide free training which gives access to a microscope for free. Besides the obvious added income, this has the hidden advantage that you do not get ‘tourists’, that is, people who just want to learn a bit or are curious but have no microscopy project. Training them is a waste of everyone's time.
- Fight against people who want you to lower the quality/duration of your training. During training, you need to give complex information to the user, likely including some theory. You need to give them time to digest this and to practice what they have learned before you move on. You need time to check that they have integrated everything. If you take shortcuts during training, it will backfire (more broken equipment, poor science). If you spend the necessary time, you will establish with your users an invaluable trust relationship. They will come to you if they have questions because your training has made them aware that you are very knowledgeable. Sylvie Le Guyader [email protected]
You have already received some excellent advice, but I'll throw in this:
1) Users are not collecting images, they're collecting data. If they don't understand how and why they generated the data (images), they cannot understand the data.
2) Discuss potential projects with users before any work is started. Not just the imaging, but the very beginning. There's no point in knowing how to collect excellent images if the work isn't done right from the start.
3) The question(s) drive the methods. Not the other way around. Sometimes this is driven by current fashions in research, but more than once I have had people who really wanted to use confocal microscopy when “plain-jane” widefield fluorescence was the better method. Or sheet-illumination when conventional confocal was the correct instrument. Some folks take convincing, and sometimes you must send potential researchers to another facility to get the right instrumentation and methods, but this is part of the job. Philip Oshel [email protected]
As to whether the training should be free, a compromise is to offer a month of free training every year. You can use this as a tool to advertise your core, and potentially increase the user base. I personally found that PIs and students normally take paid trainings more seriously, although I do not generate much more income from the training. Users pay a similar amount as the hourly user charge. ZJ Zhang [email protected]
Controls. People need to understand controls. Tissue blanks. Secondary or isotype controls. Tissue from GFP animals. “I checked this on the scope in my lab,” is not enough. Each instrument has a different response. “The postdoc in my lab already did the controls,” is not enough. Let's check how you did the prep. This discussion of collecting data, not just images, really needs to happen before a person shows up with samples. Practically, it won't. It needs to be part of instrument training. If quantification is going to be performed using the data, these methods must be developed as data are first collected. What if the plan is completely unrealistic or not proper for this type of experiment? What if there are better or easier ways that the biologists haven't thought about? Be prepared to not give researchers what they think they want, but what they need. Someone shows up at my office holding a dish of cells and announces, “I need confocal now.” They need to plan. And after talking to them, it turns out they need standard widefield, not confocal. They don't want to hear either of these messages but be prepared for this. Also, be prepared for a range of relationships from people who listen and discuss experiments respectfully and where both parties learn from each other to relationships where a person expects you to jump at their command and doesn't listen to advice. There will be a range from highly appreciative people to people who don't acknowledge you at all. There will be a range from wonderfully collaborative individuals to the most dismissive service relationship where the customer doesn't want to pay for services rendered, especially if their samples are deficient. Be prepared. Cultivate the good relationships and try to minimize the others, but you must serve all except the very few who are destructive. Billing: we are expert scientists. Try to offload as much of the administrative tasks as you can to others. Michael Cammer [email protected]
I disagree on not charging new users. PIs will send students to be trained before they need it and then they won't end up using it. If you want to promote use, charge for training but offer 2-4 hours of free use in the following week or two. Tom Phillips [email protected]
You have some good advice here and I would like to second what Phil said, great advice. I would say to always offer free training and free support. You want people to seek out training and advice without fear of cost. It will pay off in the long run with fewer damaged items and less frustrating data collection. Moreover, training does not stop with training, but instead continues throughout the core usage by the customers over months and years. You simply cannot teach everything in a single one-hour session. I would recommend doing one of the professional courses in light microscopy as it will increase your confidence that you are telling people the right things. Organize a steering committee and hand-pick the members if you can. Choose members who use the systems often (the PIs and not the actual users) and members who have some seniority at your institution. It is a good idea for a committee to be involved in decisions about the core and in large purchases because it helps to spread out any blame if outcomes are not optimal. This can especially happen when buying expensive equipment that is on the cutting edge of science. Brian Armstrong [email protected]
A lot has been said already, so just a few more points that may not have been said yet or that may have been said but I just overlooked them:
1. There are programs for starting imaging scientists. Check what Global BioImaging has to offer as they may have job shadowing programs. I also noticed some time ago a mentoring program: https://ctls-org.eu/ctls-mentors/
2. A good booking system is very helpful when running a facility. It can save you a lot of trouble when it comes to tasks like billing and reporting on facility usage. Booking systems could be a topic for a separate discussion here at the Confocal list.
3. Good IT support is a great help to an imaging scientist. I was pampered in this way at my first facility job but face a different situation at my current one. This forced me to learn a lot of IT stuff on the go, which I don't regret. Similarly, developing the imaging center website was another exciting learning journey.
4. As most others said, it is essential to cultivate good relations with the facility users and offer them a broad range of support. It is important that they don't regard the facility as a collection of equipment but rather as a seat of comprehensive microscopy expertise. Organizing courses and regular seminars can help to engage the user community.
5. Try to develop your skills in image processing and analysis. NEUBIAS and COBA have some useful resources. I have found that many users don't get the best value out of their images and even small help like a simple FIJI macro can make a big difference to their work. Many users are simply not thinking enough all that can be extracted from images (for example, just showing an overlay of green and red images rather than doing at least some simple colocalization quantification)
6. Keep track of microscope performance indicators (another big topic for separate discussion(s)). It can help detect early signs of problems as well as help find the cause of a problem when it happens. Or give confidence that there is no problem. Sometimes users tend to blame problems on microscopes when the problem is in their sample preparation.
7. There's been a discussion whether to charge for training sessions or not. Let me add my observations to this. Coincidentally, I'm in charge of 2 separate facilities which were joined into a single center. Each belongs to a different school, and each has its charging policies. One charges for training while the other doesn't. I found it an interesting case study and analyzed the records a bit, which proved my impression that there's no significant difference. The thing is that users don't pay the facility bills from their pocket and in general they are not very worried about their PIs’ grant spending. The fraction of training sessions not followed by actual usage turned out to be identical (difference of <1%) on both sides. So, I guess the decision whether to charge or not might depend more on the expectations regarding operating cost recovery. If your institution emphasizes achieving a high fraction of operating cost recovery from user fees, you better not let the training session revenue slip between your fingers.
8. I found it important to make sure new users are ready to start experiments ASAP after the training. If not (some new staff or students want to get trained on all microscopes in an initial wave of enthusiasm), it's better to postpone the training until it's relevant. Else they forget most of the stuff. That in my opinion helps to reduce the wasted training sessions more than any form of training fees. Radek Machan [email protected]
Lots of excellent advice already. I would like to add my two cents on a few points. When I started as a facility manager, the most daunting task was not the science, but the administration. Depending on whether you are taking over an existing operation, or starting a new facility from scratch, this can be a lot to learn for the average scientist. Finding a mentor is a great idea! Many of the ideas that have been posted will depend heavily on your staffing structure, and on how many instruments and users you support. If it is only you with a bunch of microscopes and a large group of users, offering free training and 24/7 personal or phone support may not be in the cards. Like others said, recovery of cost and salary expectations by the institution will also need to guide how you run your shop. Maybe a side topic, but something I came to value along the way: if you are establishing a new core, or if the core you take over doesn't have one, give it a catchy name. It helps with “brand recognition”. Good luck with your new endeavors, and don't be shy to reach out to others along the way! Elke Kuester-Schoeck [email protected]
A few minor points about training. Since charging for training seems to have “no significant difference” from free training, why not train people for free? I think that you want customers to seek out training and advice. Charging will diminish that in my opinion. Also, I always thought that imaging professionals were excited to share knowledge and to teach and train as many people as possible on correct image acquisition and the ethical representation of imaging data. Many people on this listserver have been incredibly generous with their time in the advancement of imaging science. My advice would be to never let them use their own samples during training. Have a training slide for this purpose. If you don't then you will spend most of your time talking about why their samples did not work, primary/secondary antibodies, etc. Moreover, when showing the various ways of moving the stage, adjusting a PMT or methods of focusing, you don't want to be bleaching their precious sample for minutes at a time why you elaborate on the wonderful functionality and versatility of the system. In the second training session you can show them how to image their own samples. Most people need two sessions to absorb the information of operating a confocal microscope. I think it can be good if there is some distance between the two training sessions. Consider that you are operating your own shop where the customer is always right and stress customer service. Brian Armstrong [email protected]
I agree with Brian regarding the use of a demo slide for the first training. The Convallaria test slide is ideal for this purpose. If I use a trainee's sample, then the training suddenly transforms into a discussion of their sample prep methodology and experimental results. I accompany them in their second visit when they bring their own samples. I found that it is a better opportunity to discuss often neglected controls, optimization of labeling protocols, and quantitation options. I am pretty sure that group leaders would be a bit more conservative if the trainings were not free. At the beginning of s semester, several groups often send their new students for training, and I arrange group training in such cases. Those that come with their samples later get a more focused training. It is also a good time to check to what degree the trainee remembers what I said in the first training and to polish up the overlooked points, especially those related to the instrument care. Luckily, no lens damage has happened to microscopes under my supervision, but cleaning oil from dry objectives is a monthly chore! Providing a free microscope time coupon for beginners, if they come back in 2 weeks is a good solution to encourage PIs to send their new users close to their actual use time. Ferhan Ayaydin [email protected]
Training is a serious teaching/learning/qualifying process. We do not provide free training except in extremally special cases. We provide three levels of training. Level one training covers computers and software without touching the instrument. Level one includes mandatory basic optical microscopy and digital imaging, and optionally confocal basics, image processing, deconvolution, etc. We also do remote Zoom training for level one trainings and encourage group training. Level two training covers the instrument with training slides. Group training is limited to three trainees maximum. Level 3 training uses the trainee's specimen and is limited to two trainees. After level three, if both the trainer and the trainee are confident that the trainee can run the instrument independently, will qualify the trainee to book self-service. Before that, the trainee can only book training or assisted service. Assisted service is billed at a higher rate. If a qualified user does not use an instrument for more than 6 months, they must go through a refresher session. Jingsong Wang [email protected]
Really interesting discussion. I find it fascinating to see how different facilities can adopt diametrically opposite training strategies! At our facility, we only train people with their own sample. We also ask them to prepare more samples than we can possibly image during the training so that they continue imaging immediately after the training. We never train people who have no sample, no microscopy project, or who are about to go on holiday. Concerning time generosity and how to lock users in, we choose to lock people by showing them how much we can help them. When someone contacts us, the first thing we do is set a 1 hr meeting (free of charge) where we do the following:
* Discuss their project in depth
* Explain the limitations of our systems in the context of their project
* Introduce them to the fact that everything we do in microscopy must be designed to enable/facilitate extraction of data
* Help them reformulate their scientific question in a precise way that works for quantification (for example, ‘I want to look at the green fluorescence in my cells’ can be reformulated as ‘I want to measure the intensity of GFP in a certain type of vesicle in my cells’)
* Help them define all the necessary controls (including one where we check the autofluorescence of their mounting medium)
* Give them advice and feedback about sample staining, mounting, which coverslips (#1.5 please!) or multi-well plates (low skirt please!) to use…
* Define the resolution needed to answer their scientific question, conclude which system we will train them on, and which objective(s) should be used and why
* Ask them to watch our bleedthrough video: https://www.youtube.com/watch?v=Ed1olYIjDBc and fill in a form where they evaluate the risk for bleedthrough between the fluorophores on their sample when imaged on the microscope we will train them on. They must bring the form with them on the first day of the training
We do this with everyone who contacts us, regardless of whether they will use our facility or not. I think that it is generous, and we do enjoy the brainstorming aspect of these meetings. The consequence is that people immediately see that they can rely on our expertise and that we set from the start a team relationship with our users. I would say that at least 95% of our users just follow all the advice we give them during the first meeting. The training is then geared at giving a boost to their microscopy experiments where they not only acquire relevant data, but where we troubleshoot their sample preparation and once more discuss how to extract data. I believe that the only way to validate a teaching/training strategy is to first define what the learning outcomes are (what MUST the student have learned at the end of the training) and to then to test if they have reached the goal. I would guess that one of the main differences between facilities is that the learning outcomes of the training are very different. Additionally, if they are not clearly formulated at the start of the training, it might be difficult for both trainee and trainer to reach them. I would also guess that trainees are very rarely tested for having fulfilled the learning outcomes. While the “Do Not Break Anything” learning outcome is probably a must, it is not sufficient. Here are our learning outcomes for everyone who will acquire images and will need to adjust settings themselves (most users) regardless of which type of sample/scientific question they have:
* How to book time and rules at the facility
* How to avoid damaging the equipment
* How to start the machine safely
* How to safely find your sample without bleaching it (https://www.youtube.com/watch?v = 3CdiMEHlF_Y)
* How to operate the software to combine all the dimensions (xy, z, t, lambda, multi-well) required by the scientific question
* How to define the xy resolution needed from the scientific question and to check if the objective can deliver this resolution
* When is it necessary to match sampling (Nyquist) with the resolution of the objective and how to do it
* Same thing for z resolution and sampling if the project needs z-stacks
* How to save (format) and export data in a safe way
After the training, we have a meeting with the image analyst where we start designing the analysis pipeline using the images acquired during the training. We are currently writing a paper about training users at imaging facilities. I would be delighted to collect (online or offline) the main points that according to you, your trainees MUST integrate in their practice at the end of your training. Sylvie Le Guyader [email protected]
Afterthought on training and why we insist on real samples; many of our customers/clients/users are new to biology. This is an opportunity to teach basic cell biology. For instance, a new student is rotating in a lab that has them looking at a novel protein. We can say things like, look, it appears to be aggregating in the Golgi region. Then they ask, how can you tell what is the Golgi region? We explain based on morphology, but that this could be considered conjecture, so how more definitive tests could be done. And, more importantly, what does this mean from a functional standpoint. Also, on a few occasions we have interceded in situations like those described in this paper: https://www.jbc.org/article/S0021-9258(17)49822-X/fulltext. People say they've stained for something, but clearly, they are labeling something else. Or they say they've stained something with Alexa 488, and it turns out maybe they did, but someone gave them eGFP cell lines for something else. So, QC and teaching. Michael Cammer [email protected]
Dear All, Thank you for your recommendations and tips for new core facility managers. I value them and note them for easy access. I am more enlightened today because of your brilliant contributions. Thank you. I have registered on the global bioimaging website and the bioimaging mentor's program, and I will watch out for training opportunities. Alicerita Eseola [email protected]
Fee Structure for Consulting/External Use of a Core Facility
Microscopy Listserver
Dear Microscopists, my university is trying to develop a fee structure for consultation and/or external use of our FE-SEM/EDS lab that ideally provides enough funds to pay for maintenance of the instruments, eventually provide replacement funds when instruments age beyond end of life, and feed the operator (probably me). Question: What are typical external rates for SEM/EDS analysis? Thank you for any advice you can share. Kurt Friehauf [email protected]
If you are trying to charge users on federal grants, then you have to work with your “research accounting” department/people at your university to set the on-campus rates. After that, you can set your off-campus rate, which at minimum is 15% higher, but should be “market rate”. There are federal guidelines. I hope this message does not set off the typical fire storm on rates. Jim Quinn [email protected]
Broadly speaking, we take the cost of the SEM service contract + cost of the EDS/EBSD service contract + cost of about 100 hours of a staff member or technician for maintenance and upkeep + cost of consumables + about 5% contingency and divide by 35 hours/week X 50 weeks/year. Depending on the machine, this comes in around 80-150 US$/hour. We aren't allowed to charge different customers different rates, and we can't escrow money past September 30th to buy a new machine in a future year, so your milage may vary. Chad Parish [email protected]
Our facility is also looking into creating external rates. I recommend reading this article: “Best Practices for Core Facilities: Handling External Customers” (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3605920/). Guideline 4 is important; If there is a commercial vendor with comparable services within a defined radius of your institution, then you may not be allowed to provide services to COMs that are below that rate. You may provide your service at comparable or higher rates, however. Your institutional guidelines on this may vary in terms of the distance restriction. Elizabeth Miller [email protected]
How are Customer Codes in Relion Added?
3DEM Listserver
Is it possible to add customer codes or scripts to Relion? For example, I would like to program for extracting a region of a map for local refinement and integrating the codes into Relion. Thanks. Sunyeping [email protected]
If the task is independent from RELION, you can call your own program/script from the RELION pipeline as an external job. If the task has to be executed every iteration during refinement (for example, density modification / filtering), you can use the external reconstruction feature as a hook. SIDESPLITTER uses this mechanism. If you need to modify RELION's behavior, you have to hack the source code. Your mileage may vary. Writing a new program with RELION's library functions is simple; write your own program and put it in the src/app folder. src/app/xxx.cpp will be compiled as relion_xxx. Tsakanori Nakane [email protected]
Secondary Electron Detector Scintillator Phosphorous Film
Microscopy Listserver
Dear All, I am looking for a procedure or method to recover phosphorus film from the secondary electron detector scintillator. Does anyone have literature on this? Thanks Antonio Carlos Joaquim [email protected]
May be easier to just order. https://www.tedpella.com/Scintillators_html/Scintillators.aspx#P47 David Hull [email protected]
My buddy used Mop & Glo® (random cleaning compound) as a binder to recoat his Everhart-Thornley detector. Made a thick paste and formed it on the end. It was thick enough to sand off, probably with high grit sandpaper (he tried this after suspecting the coating was too thick and thus reducing his signal brightness/quality). He said the p47 vial mentioned before would be a lifetime's worth of phosphor for recoating detectors. Nathan McCorkle [email protected]
We've used M.E. Taylor to recoat our SE scintillators in the past. Alan Stone [email protected]
Here is a recipe for coating low-energy electron diffraction (LEED) optics. It must be adapted as we used it for a 120-150 mm screen, and you need it for a 10-25 mm scintillator. But it provides some hints, like the use of sodium silicate as cement to hold the fluorescent powder together. It is important to put an Al layer on the surface at the end to make it conductive. The difficulty will be to find the right fluorescent powder in a small quantity, and to put the right thickness (signal versus absorption). You must also verify that the fluorescent color of the powder is compatible with the spectral sensitivity of the PM. Most powders provide green light, but I've seen some which give blue.
Fluorescent viewing screen for LEED optics – 120mm diameter screen
Bath A: 1 g (NO3)2Ba in 2.6 L distilled water
Bath B: 2 g fluorescent powder (Willemite (Zn2SiO4), Cathodix 602, Massiot Fluor H913 in 800cc distilled water. The powder must be added carefully, to avoid lumps, with a magnetic shaker. This solution must be filtered (100 microns or less filter) to remove lumps, and ultrasonicated for 10 minutes. Add 100 cc of ordinary sodium silicate.
1. Clean the screen with KOH/alcohol solution and rinse with distilled water.
2. If the glass has no conductive coating it is good to put a coat with a fine layer of gold (10 nm).
3. Place the screen in a 3-4 L beaker so the screen is a few cm from the bottom. It's better to use a glass support than a metal one. The support must be smaller than the screen. Typically, a tripod made from welded glass rods works well. The screen must be precisely horizontal.
4. Mix the two solutions. Agitate it hard and pour it in the beaker. The fluorescent powder will sediment in 1 or 2 hours.
5. Slowly siphon the liquid part very carefully with a glass or rubber tube. Be careful not to swirl the mixture.
6. Carefully place the beaker in an oven and warm it progressively from 50°C to 150°C for 15 to 20 hours. The screen must be precisely horizontal. If possible, it is better to put the beaker in the oven after step 4. You can then siphon in the oven. For LEED or RHEED screens, we put a layer of aluminum on the coating to avoid charge accumulation. I don't know if it is necessary for EM. And I don't know if the granularity of the coating is fine enough for EM. For LEED or RHEED, the size of the spots is at best a few tens of milimeters. Of course, never touch the surface. This recipe is from Werner Espe's “Hanbuch für Physik”. Jacques Faerber [email protected]
Please see NR Comins et al., J Phys E: Sci. Instrum., Vol. 11, 1978. Preparation and evaluation of P-47 scintillators for a scanning electron microscope: https://doi.org/10.1088/0022-3735/11/10/021. Henrik Kaker [email protected]
Advice on TEM/EDS Detection Thresholds
Microscopy Listserver
My inquiry is concerned with understanding the function of the silicon drift detector (SDD) and the associated hardware/software subsystems. For this reason, please neglect sample issues where feasible. These samples were in solution, so I used a glass pipette to place a few drops on the grid (usually formvar) and placed them in the desiccator overnight to allow them to dry before placing them in the TEM for observation. The reason for my question is that I encountered a signal for atomic oxygen that ultimately was not detected by our Oxford X-Max 65T. This is a windowed SDD. The two complications to this are as I watched the AZtec scan progress I observed a momentary identification of oxygen. This apparent contradiction caused me to contact Oxford for assistance. Oxford instructed me to enable the elements I expected to see in the sample. I think the motivation for this is that informing the detection system what you expect to see helps the characteristic peak fitting routines they use. A colleague learned of this issue through discussions within the department and came to examine the data. His conclusion is that oxygen was present. Oxford and the X-Max 65T say no as the signal was not above the threshold to be confirmed. Lastly, I should add I did not receive formal training on this instrumentation. I was hired nearly a year after it was commissioned and signed off. Oxford was good enough to provide me with phone support when necessary. I just need a better understanding of the issues involved. Rick Van Camp [email protected]
Light element EDS is problematic in general; detection limits are poor primarily due to a low fluorescence yield. The issue in your case sounds like a statistical one. The counts in an EDS spectrum are described by the Poisson distribution. In the Poisson distribution, the standard deviation (sigma) is equal to the SQRT (counts). This means that the measured counts may vary even when there is no peak present. So, a statistical test is used to say with some certainty that the counts seen did not arise by chance. The standard measure is a 3-sigma test, that is, the peak counts measured must exceed the background counts by 3-sigma. This gives a 99.7% confidence that the peak is real. I suspect that in your case, the counts didn't quite meet the statistical criterion set by Oxford, especially since the label appeared only momentarily. The counts may have been right on the edge of “detectability”. Henk Colijn [email protected]
Your question mostly deals with software rather than hardware. It would apply equally well to SDD or Si(Li) systems, with windows or without. Those matters could be the subject of another thread. I agree with Hendrik's comments. Noise in the background can lead to the appearance of a peak when none is significantly present. It is common for peak identifications to appear and disappear during collection. I work with materials, and it is common to see S or Cl pop in and out. My eye (and gut) can often tell there is a faint peak there, but the software isn't sure until the counts increase, and the statistics improve. (Moral: More counts are generally good.) But then there are the practicalities of implementing these principles in software. (Disclaimer: I have an Oxford Aztec system. I am generally quite pleased with it, but it is not perfect. Probably no system is.) I mentioned S and Cl above. They are at higher energy and relatively free from complicating peaks and structure. For these, it is usually just a matter of counting longer. Unfortunately, things get busier and more complicated at low energies. There are lots of elements close together as well as other low-end structures like Oxford's strobe peak. We look at a lot of polymers, so we have strong C and O peaks. Often, we also have N (as in Nylon), but that peak can go unidentified even though it is most certainly present. There seems to be some practical difficulty in automatically identifying that peak when it is between two strong peaks. Our version of the software has a step named “Confirm Elements”. That is not just an option but should be considered a necessity. Automated peak ID software has greatly improved over the years, but it is not perfect. One would be foolish to take the automated output as the final answer. It really does need to be confirmed by more intelligence than that encoded in the software. You said Oxford suggested manually predefining elements as present. That is a reasonable practice. I routinely instruct our users to employ this in cases where they might have a small amount of an element, particularly if it is in the neighborhood of a major element. For example, someone working with ITO will find the indium peak to be quite small on the shoulder of a very strong tin peak structure. The indium will probably not be identified automatically. If you carefully examine a fitted spectrum without indium, you should notice the indium intensity. (I toggle the indium on and off to see how the fit changes.) Therefore, I have no hesitation about including the peak and letting the quantification software determine the concentration. If it is less than 0.1%, I may discount it, but Oxford does a quite good job with their peak shapes. After that, I would encourage you to take advantage of training available through Oxford. I don't know what training classes look like now. I remember several days of training on a Tracor-Northern TN-2000 back in 1980 (then Noran, now ThermoFisher). It was time well spent. I learned a lot about EDS. It was also a nice opportunity for networking. Warren Straszheim [email protected]
As someone who worked with making ITO thin films and quantifying the resultant film, spot on. You can watch the indium peak appear and disappear even with high count values. Some knowledge of the source material under the beam to feed the algorithm improves confidence in the analysis. Jason Sabeliaree [email protected]
I would add that even if the peak is not 3-sigma above background, it may still be 2-sigma (or 2.x-sigma) above background. In which case the detection might not have 99% confidence, but only 95% (or whatever) confidence. Replicate measurements, and a t-test can also be useful in such cases. By the way, in the Poisson distribution the standard deviation only approaches the sqrt (counts) at high (enough) count rates. But we all assume this relationship for ease of calculations! John Donovan [email protected]
To amplify Warren's comment about auto peak ID, Dale Newbury and Nick Ritchie have discussed auto Peak ID in several articles. I seem to remember that a few years ago Dale said that the auto Peak ID had about a 3-5% error rate on major constituents. “Elemental misidentification typically occurs in a few percent of attempted peak identifications for major constituents, with the frequency of mistakes increasing significantly for minor and trace constituents and especially in cases where peak interference occurs…” (DE Newbury and NWM Ritchie, J Mater Sci 50 (2015) https://doi.org/10.1007/s10853-014-8685-2). One that I saw early in my career was whenever I looked at stainless steel, the EDS system would happily tell me I had Promethium (overlap with Cr). Auto Peak ID has improved greatly since I started, but it can still be wrong. Henk Colijn [email protected]
In fact, Dale often referred to these qualitative analysis issues as “blunders” in microanalysis! John Donovan [email protected]
I am not sure that the problem has much to do with software, detectors, shapes, etc. Formvar contains oxygen and it is normally seen in EDS spectra in the TEM. My guess is that the sample is too thick or close to a grid bar. Krassimir Bozhilov [email protected]
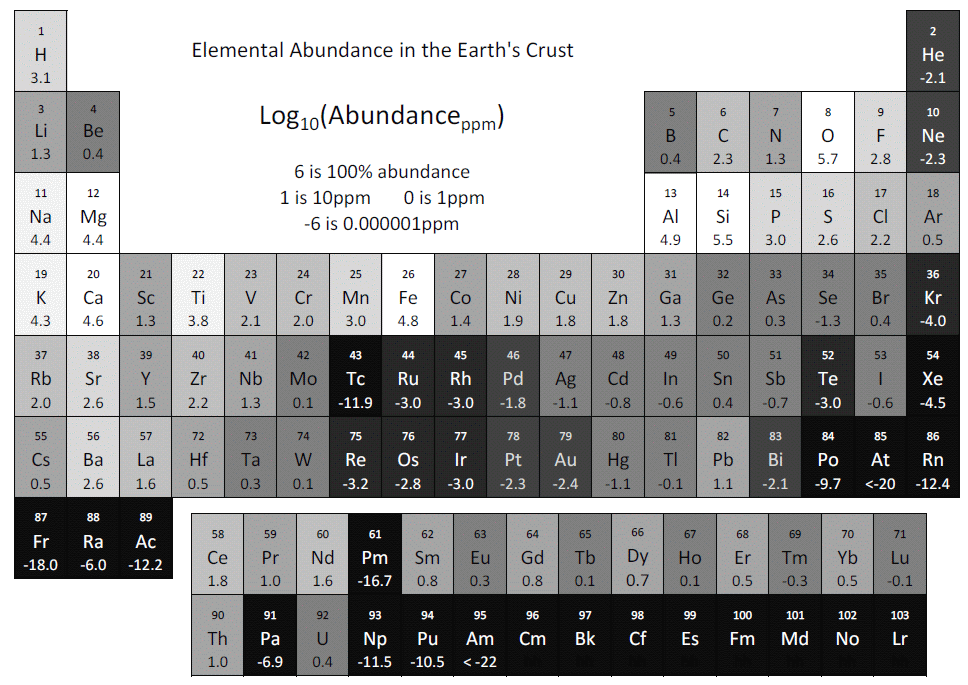
When showing students how to do peak identification, I now use this: http://DoL1.eng.sunysb.edu/EDS/earth-abundance-table.gif. The table is shaded grey to indicate abundant and rare elements, on a Log10 scale. Oxygen is 461,000ppm (46.1%), hence Log10(461000) = 5.7. Promethium/Pm is 0.00000000000000002ppm, hence Log10(0.00000000000000002) = -16.7. It is from the compiled values (column CRC[7]) at: https://en.wikipedia.org/wiki/Abundance_of_elements_in_Earth%27s_crust, from the CRC Handbook. My belief, all elements <-6 should be auto-excluded from any manual or auto peak identification: Tc -11.9, Po -9.7, At <-20, Fr -18, Ac -12.2, Pm -16.7, Pa -6.9, Np -11.5, Pu -10.5, Am <-22, Z>96, and inert/rare gases He, Ne, Ar, Kr, Xe, Rn. Jim Quinn [email protected]
Hopefully this can be configured in the software. I know we can in our version of Aztec. I generally concur with your list, but I have found real Ar in some sputtered samples. Some of it was in an Al sample, and no, it was not a sum peak. I suppose we could also start finding Xe nowadays. It has been fun getting familiar with more of the periodic chart. Warren Straszheim [email protected]
Your chart looks to be quite useful in a university setting where there are a lot of relatively inexperienced users. As Warren and Chad pointed out, there are cases where we *do* need those less-common elements, but for many users it can reduce the chances of an off-the-wall misidentification. Henk Colijn [email protected]
96-Well Plates for High Numerical Aperture Imaging
Confocal Listserver
Hello, everybody. I'd like to know if you have some recommendations for 96-well plates (well not necessarily 96) that would be suitable for high-NA imaging. Ideally the plate would have a very homogeneous thickness, of the order 170-190 mm, and have no skirt so all wells can be imaged with high-NA objectives. The well-plates we recommend at our facility are the Perkin Elmer PhenoPlate 96], but we would like to have options. Thanks! Nicolas Chiaruttini [email protected]
I did a search a while back and here's a list of plates I found. All have low skirts and plate bottoms compatible with oil imaging: Corning 4517, Greiner 655891, Greiner 655866, Perkin Elmer 6055302 (and related; this is a PhenoPlate™), Brooks MGB096-1-2-LG-L, and Nacalai USA 5882-096. The PhenoPlates are very good, particularly the 384-well ones. Kurt Thorn [email protected]
At our facility we use 96-well plates, dishes, and imaging chambers from Zell-kontakt (https://zell-kontakt.de/). Gabriela Imreh [email protected]
What about the ibidi chamber 8 well glass bottom? https://ibidi.com/glass-bottom/252--slide-8-well-high-glass-bottom.html These are recommended for TIRF microscopy and therefore are used with High NA objectives. Fernanda Gárate Marín [email protected]
Personally, for high NA work I have had good experience with the Cellvis plates. Despite the skirt height we have no issues accessing the outer wells of the 96-well format with oil lenses on both our NIkon and Zeiss scopes. https://www.cellvis.com/_96-well-glass-bottom-plates_/products_by_category.php?cat_id = 11. Perkin-Elmer also makes a decent plate, but the Cellvis are my favorites. With high NA work (.75 and higher), the challenge really becomes maintaining focus across the individual wells and across the field-of-view. With the short depths of field of the 1.3-1.45 lenses, small variation across the plate is difficult to overcome even with amazing laser autofocus. I've had terrible luck with warped wells with the Greiner plates and with Nunc and am hesitant to even try automated imaging at higher numerical apertures with those plates because it almost always ends in frustration and heartache. I'm happy to speak more off-list about this - for better or worse I've had a lot of experience troubleshooting screening using oil objectives. Cassandra Rogers [email protected]
ibidi also has multi-well plates (https://ibidi.com/23-multiwell-plates). We recommend these to our core facility users because with many others we ran into the problem of a warped bottom making focusing difficult, while these seem to have a flat bottom throughout. The website of each product has a “Get your free sample” button, so that may be worth trying. Steffen Dietzel [email protected]
I use Cellvis plates available for standard number 1.5 glass, high precision 1.5 glass, and 1.5 equivalent plastic (presumably cyclic olefin copolymer). I have tried most manufacturers’, and these are the best I have found. Interestingly they are also the cheapest by quite a bit. The 96-well plate is cheaper (and better quality) than ibidi or Nalgene plates with 6, 8, or 12 wells. For a small experiment just use the 2, 6, or whatever desired well count you need in the 96-well. Second point to consider is your hope to utilize all the 96 wells. I recommend against this since there are edge artifacts. I never use the outside row/columns. This leaves the middle 60 wells. A paper on the edge effect is M Mansoury et al., Biochem Biophys Rep 26 (2021) https://doi.org/10.1016/j.bbrep.2021.100987. Also, consider skipping the outer 3 rows/columns in the 384-well format. A final note is to fill the edge wells with water/buffer to provide extra humidity for the plate. Ryan Schreiner [email protected]
How Much More Light can be Captured by a 4X than a 2X Objective?
Confocal Listserver
Dear all, I just received several images that are most curious! The same HCS plate with fluorescence-stained HeLa cells was imaged twice with the same instrument, once with a cheaper 2x and once with a more expensive 4x objective. The curious part is that the signal-to-noise ratio of the 4x images is better than that of the 2x images, by such a margin that it is hard for me to believe. The 2x images are mostly just noise, whereas in the 4x images I can clearly see the cells! This is consistent over three channels. Admittedly, I do not have many details about the hardware (yet), so my question is rather generic: Just based on this little information that the SnR at 4x is much better than that of the 2x, would people think there is another, hidden problem in the 2x images? Maybe Köhler alignment was poor or there are other problems in the light path? Or is it possible, and likely, that the 4x is so much better? All the best, and happy imaging, Mario Emmenlauer [email protected]
It could be as simple as the numerical aperture of the 4x objective is better. Not to pick on any company, but Leica sells a 2.5x/0.07 and a 4x/0.13. Using the fluorescence brightness calculations (found here: https://www.olympus-lifescience.com/en/microscope-resource/primer/anatomy/imagebrightness/) I calculate a nearly 300% increase in brightness by going to 4x. Douglas W. Cromey [email protected]
The NA of the lenses is what matters. Assuming 2x higher NA of the 4x lens than the 2x lens, the 4x lens will focus four times as much excitation light per square micron (assuming overfilled back aperture), and collect four times as much fluorescence per square micron, but it will distribute the light into four times as many camera pixels per square micron (of the sample), leading to an overall 4x improvement of per pixel SNR (but 16x improvement of per cell SNR). Definitely, more info is needed to give a clear answer. Zdenek Svindrych [email protected]
Oh my, I was not aware that NA can differ so much. That's quite interesting! Then I'll assume everything is ok with the hardware, and I learned something new today. Thanks! Mario Emmenlauer [email protected]
A bit tangential, but if you want to do low magnification epifluorescence, these objectives work very well: https://www.olympus-lifescience.com/en/objectives/xlfuor/. Per the NA intensity app Doug pointed out, this would give a 16x increase in epifluorescent intensity over the standard Leica 4x objective. We use it for calcium imaging across a 4.75 mm x 4.75 mm FOV and get very good SNR with low-excitation intensity (low enough that we can do 10 Hz imaging for 1 hour without any appreciable photobleaching). Benjamin Smith [email protected]
Seeking Advice Concerning Genetically Fluorescent Proteins for Multiphoton Imaging
Confocal Listserver
We are looking for a better selection of four genetically encoded fluorescent proteins for use in imaging live tissue. For approximately 15 years we have been using CFP, GFP, YFP, and tdTomato or another red probe, typically exciting all at once at 930 nm. This works, but separating the signals requires significant cross-subtraction and works best when each probe is expressed by a different cell type. We now have a system with two lasers, one of them an Insight that can be tuned out to 1300 nm. Therefore, we can scan sequential by line, for instance exciting at 850 and 1100 nm. The red signal is far brighter exciting at 1100 nm (and brighter still at 1150 nm, but then we also get 2nd harmonics in the red channel). We are curious whether anyone has a suggestion of different probes, perhaps a combination with something in the near IR, so that the colors can be better distinguished? Thank you! Michael Cammer [email protected]
I can comment on one aspect of your inquiry. The red proteins our users have successfully imaged are tdtomato and mCherry. mCherry has an emission max at 610, so you should not get into trouble with second harmonic generation (SHG) in the same channel. At least it was not a problem here. mCherry comes up in the near red, like AlexaFluor 594 or Cy3.5. Steffen Dietzel [email protected]
Another option is to tighten up the bandpass filters in your system to be more selective. This will reduce the signal but may improve the crosstalk situation. Options for transgenically encoded fluorophores are still somewhat limited. For discrimination of SHG, I use an extremely narrow bandpass filter, around 10 nm in bandwidth. Tuning the laser 10-20 nm will reveal or conceal the signal, so it is a quick-and-easy confirmation that the signal I am seeing is indeed SHG and not fluorescence. This trick can be used with any (relatively) narrowband non-linear effect. Craig Brideau [email protected]
Bake-Out Procedure
3D Listserver
Dear all, we recently received our Gatan Elsa 698 holder. I would like to gather some recommendations for daily use and bake-out procedure. What temperature do you use for bake-out, how long do you bake-out for, are there special maintenance procedures, what are the signs of deterioration, and after what period would they occur? We use the holder both for room temperature and for cryo-measurements. The manual refers to the Gatan pumping station, but ours was delivered with a Pfeiffer pumping station. Thanks a lot for your time! Best, Tim Gruene [email protected]
I am not familiar with current cryo holders but have some old info to share. The 626 Gatan cryo holders used zeolite and now, to trap water when cooled down. This improves the vacuum around the holder Dewar and improves temperature stability, which in turn improves drift behavior in the microscope. Baking out on the 626 meant a 750mA starting current to get to 100oC. When 100oC was reached, the current was adjusted to maintain 100oC. As the zeolite or carbon gasses out, vacuum improves and less current is needed to maintain temperature. So, the current is the spec to watch. On the 626 I settled for 450mA. I figured this out some twenty years ago in the Philips factory: at the time bake-out time was fixed to a few hours by the Gatan controller and had to be done once a week according to the Gatan manual, but I noticed different behavior on all the new holders I got from stock. We, however, didn't know when the holders came into the warehouse at Philips. Multiple bake-out cycles, while measuring vacuum, improved holder performance, and this correlated with the current-drop needed to maintain 100oC during bake-out. So, the bake-out time was made a user setting on the Gatan controller. Once a 626 holder reached 450mA after bake-out, it was fine. But maybe I am a dinosaur on this, so I am happy to be corrected. Wim Hagen [email protected]
We use Elsa 698 with the Gatan 1905 controller and 655 pumping station. The bake-out temperature is kept at 100oC, never exceeding it for more than a few degrees. We usually perform overnight bake-out once a week, but we pump out the holder Dewar after each cryo-day. I think that it is ok for the holder to perform bake-out more often in case of extensive sample exchange. You need to do the holder bake-out if:
1) The holder fails to reach the -170oC temperature within a reasonable time. In our case ~45-50 min inside the pumping station, but may be different for your holder.
2) The holder cannot keep the temperature cold enough during operation.
3) Frost or condensate form on the holder Dewar.
4) The holder was not used for a couple of weeks.
If the holder was not used for a longer time, or if overnight bake-out does not solve the temperature problem, 48 to 72h bake-out times should be used. Andrei Moiseenko [email protected]
The modern Gatan controller includes a manual mode that allows to set the current. The user-friendly menus show the temperature, but most likely the controller itself uses current controlled by temperature. Tim Gruene [email protected]
Hi Andrei, thanks a lot for sharing these details! I was mostly curious about frequencies of the various steps (just pumping / pumping and baking), and the temperature. My former PhD student, Julian Wennmacher, would bake out zeolites at much higher temperatures, and I would have guessed 150oC. So, it is good to know that 100oC is recommended. Tim Gruene [email protected]
For what it is worth, I seem to recall being told many years ago that over time the construction of cryo-holders changed (perhaps the type of neck epoxy used?) and the recommended bake temperature changed with it. Of course, there are always the “more is better” folks, but I think it best to stick to the temperature recommendation and we use 100oC. Dennis Winkler [email protected]
I agree 100% with Dennis. Basically, baking any Gatan cryo-holder, for example, 626, 636, or Elsa 698 above 100oC, could mean expensive repairs for the holder. There are components soldered to the internal thermal conductor that are likely to be damaged if the holder is baked at elevated temperatures. I would stick with the 100oC bake-out temperature, unless of course the Elsa user manual specifies otherwise. Tom Schmelzer [email protected]
Bone Ultramicrotomy
Microscopy Listserver
I have several projects requiring TEM of decalcified mouse bone. The samples are processed using standard protocols and embedded in Spurr's resin. Prior to ultrathin sectioning, I take 0.5 μm semi-thick sections of a large field of view so the researcher can select the region of interest to target for TEM. These semi-thick sections are cross-sections through long bone (tibia, femur), and they always turn out horribly wrinkled. I've tried using a heat pen to flatten them. I've also placed the sections on a drop of water on a slide and covered with a petri dish lid coated with xylene hoping the vapors will relax the tissue. These methods work for all other tissues except those rich in collagen such as bone and tendon. The tissues do tend to flatten better when I cut the bone parallel to the long axis (I'm guessing most of the collagen fibers are oriented in that direction), but the researcher desires cross-sections. Does anyone have a trick to flatten these troublesome sections? Shannon Modla [email protected]
Is the mouse bone in the semi-thick sections surrounded by empty resin? Folding often occurs when drying as the tissue swells more than the surrounding resin. If so, trimming off the empty resin might help. Ralph Common [email protected]
This is a common problem for cross-sections of mammalian hairs in TEM. Some folding is unavoidable. Increased thickness can help reduce folding, but at the expense of resolution. In the case of hairs, the problem is due to the lack of infiltration of resin so (as Ralph Common said) it seems like a resin-tissue difference. I'm presuming decalcified bone infiltrates ok, but anything that improves infiltration or fixation following decalcification may help. A small trick we use is chloroform vapor (just a tiny drop on a loop) held above the section as it floats on the knife boat. It's like the xylene trick but with an even less pleasant chemical. I have no idea if it will work with collagen but might be worth a go. Duane Harland [email protected]

{kind=link}