


Typical brain development follows an expectant rate during fetal and early neonatal life. The process of typical brain development has been well described at the neural circuit and gene expression levels (Allen-Institute, 2010; Thompson & Nelson, Reference Thompson and Nelson2001). Development is largely driven by genes that are independent of environmental stimuli, allowing the brain to develop relatively normally in spite of a wide range of environmental conditions, a concept particularly relevant to the diathesis–stress model of psychopathologic etiology (Monroe & Simons, Reference Monroe and Simons1991). It is likely that this “clocklike” development assures a fundamentally similar outcome across a wide range of human experiences. Nevertheless, it is clear that experience plays a role in brain development and that the impact of experience is greater during periods of rapid growth and differentiation (Cross-Disorder Group of the Psychiatric Genomics Consortium, 2013). The remarkable overlap among genes identified as risk factors for multiple disparate psychopathologies suggests that a common fundamental grounding leads through environmental factors to multifinality of the behavioral phenotype in adulthood (Cross-Disorder Group of the Psychiatric Genomics Consortium, 2013).
The impact varies based on timing of the experience because the brain is not a homogenous organ. Rather, it is composed of different regions, cell types, and processes (e.g., myelination and neurotransmission). Each has a different developmental trajectory and therefore peak period of rapid growth and differentiation. For example, the hippocampus's peak period of development starts in the third trimester and proceeds through the first 18 months. Its peak is well before that of the prefrontal cortex, which matures later postnatally.
This differential development timing becomes crucial when determining the effect of a fetal environmental event on a circuit that relies on the integrity of both structures (e.g., the ventral tegmental area [VTA] loop). This neural circuit integrates multiple behaviors and dysfunction of it through imbalance of its tonic hippocampal input and phasic frontal lobe input has been implicated in the etiology of schizophrenia and drug addiction (Esmaeili & Grace, Reference Esmaeili and Grace2013). To illustrate the differential effect of timing of a fetal environmental event, we can consider what occurs to its construction and function in a preclinical rodent model when a nutritional substrate that is crucial for neuronal growth and differentiation (i.e., iron) is deprived in the fetal and newborn period when hippocampal, but not frontal lobe, development is particularly rapid. In this illustrative case, early life iron deficiency results in an adult animal with significant hippocampal, but minimal frontal lobe, impairment. The effect at the circuit level is complex and results in hippocampal disinhibition, which allows for greater dominance of frontal lobe inputs into the VTA loop (Schmidt, Alvarez, Grove, Rao, & Georgieff, Reference Schmidt, Alvarez, Grove, Rao and Georgieff2012). Consequently, the adult shows greater cognitive flexibility on set-shifting tasks than always iron sufficient controls in spite of also demonstrating rather poor declarative memory. This example emphasizes the importance of both timing and duration of early life environmental events on subsequent adult functioning.
Fetal environmental effects can cause acute functional changes to the brain that return to normal once the environmental effector is removed. In the case of short-duration environmental effects, the long-term effects may be minimal if no critical periods of development were missed and if environmental homeostasis can be reestablished. However, all of the early life environmental effects that are discussed in this article have been shown in clinical studies and preclinical models to cause disruption of adult function, suggesting important long-term changes to brain anatomy and the molecular biology of synaptic plasticity gene regulation.
From a societal standpoint, the cost to society of early life adverse environmental events stems from the long-term effects, manifested as loss of education and job potential, medical costs for treatment of mental health issues, and loss of opportunity to improve socioeconomic status. Policies and practices that do not ensure optimal early life environmental health represent a failure of a society to invest in its next generation. The adage “how are the children doing?” takes on full transgenerational biological meaning via mechanisms underlying the developmental origins of adult (mental) health and disease (DOHaD) paradigm. In this article, we review three major categories of early life environmental disruptions that confer long-term risk to the mental health of humans: fetal alcohol exposure, teratogen exposure, and nutritional deprivation.
It should be noted that differentiating fetal from early postnatal life effects is relatively arbitrary on the following counts. First, it may be difficult to separate exposures into such defined epochs. Nutritional deficits accrued by the fetus carry forward into postnatal life. For example, iron is accreted primarily during the last trimester and is utilized by the fetus to support regional neural differentiation, myelination, and monoamine neurotransmitter production (Lozoff & Georgieff, Reference Lozoff and Georgieff2006). Fetal iron deficiency results in acute recognition memory failure, slower speed of neural processing, and a disengaged newborn infant (Amin et al., Reference Amin, Orlando, Eddins, MacDonald, Monczynski and Wang2010; Siddappa et al., Reference Siddappa, Georgieff, Wewerka, Worwa, Nelson and Deregnier2004; Wachs, Pollitt, Cueto, Jacoby, & Creed-Kanashiro, Reference Wachs, Pollitt, Cueto, Jacoby and Creed-Kanashiro2005), symptoms that are completely consistent with the known biology (Lozoff & Georgieff, Reference Lozoff and Georgieff2006). However, low iron stores at birth due to reduced fetal accretion also results in reduced substrate for rapid postnatal brain growth and differentiation. The relatively slow transfer of postnatal dietary iron across the blood–brain barrier results in an extended period of additional postnatal brain iron deficiency and thus confers a risk to more, postnatally developing circuits. Similarly, teratogens can accumulate in the body during fetal life and thus affect fetal brain development acutely. Postnatally, those stored teratogens can continue to damage developing structures and alter gene expression. In contrast, fetal alcohol exposure clearly ends with delivery of the baby from the alcohol-consuming mother, thus removing any risk of postnatal exposure.
Second, the DOHaD hypothesis, initially known as the Barker hypothesis, initially focused on adult cardiovascular risk as a function of restricted fetal nutrition and growth (Barker, Reference Barker1997). Barker's conclusion that “fetal programming” accounted for the increased risk of hypertension, type 2 diabetes, and cardiovascular in the elderly instigated the search for fetal factors that mediate the effect. Findings since the original reports indicate that the effect is not solely restricted to fetal life, but that the transition from fetal to neonatal life happened to provide a convenient “cut point” to study, that is, a time when a human goes from one nutritional environment (i.e., intrauterine) to a potentially radically different one (i.e., extrauterine). A major refinement of Barker's “fetal programming” hypothesis came in the early 2000s when Gluckman and Hanson (Reference Gluckman and Hanson2004) demonstrated that the predictive value of fetal growth restriction for adult heart disease was much greater if one considered the growth rate of the individual postnatally during the first months of life. Rapid postnatal weight gain after intrauterine growth restriction (IUGR) exacerbated the risk in adulthood (Gluckman & Hanson, Reference Gluckman and Hanson2004), and it was postulated that any jarring change in environment early in life induces stress responses as indexed by altered hypothalamic–pituitary–adrenal axis regulation and pro-inflammatory responses as indexed by elevated pro-inflammatory cytokines. Both responses are injurious to developing organs such as the heart and blood vessel systems. If IUGR fetuses grew slowly postnatally, their risk of cardiovascular diseases was mitigated. The findings raised the possibility that a developmental window exists when metabolic set points are set for the life span, likely through epigenetic mechanisms.
Subsequent studies in premature infants, who frequently undergo severe growth restriction postnatally, also demonstrate an increased risk of central adiposity, early life hypertension, and insulin resistance. These infants would have had their growth restriction followed by rapid catch-up growth in the same developmental window as Barker's IUGR subjects (i.e., 24–40 weeks postconceptional age), but all of the events would have occurred in the extrauterine environment. The postnatal duration of the sensitive period for setting metabolic set points is unknown. Data from internationally adopted children demonstrating increased central adiposity and relative stunting suggest that the period may extend through the first year of life. Although the birth history of most of these children is poorly documented, it is clear that they undergo a period of malnutrition, stress, and growth restriction until placement (Johnson et al., Reference Johnson, Miller, Iverson, Thomas, Franchino, Dole and Hostetter1992, Reference Johnson, Guthrie, Smyke, Koga, Fox, Zeanah and Nelson2010). Multiple endocrine mediators of growth and development are disrupted (Miller et al., Reference Miller, Kroupina, Mason, Iverson, Narad, Himes and Petryk2010). Adoption into a resource-rich home typically stimulates rapid catch-up growth, consistent with the abrupt change in environmental conditions described by Gluckman and Hanson (Reference Gluckman and Hanson2004), and accompanied by altered hypothalamic–pituitary–adrenal axis regulation (Gunnar & Quevedo, Reference Gunnar and Quevedo2008; Kertes, Gunnar, Madsen, & Long, Reference Kertes, Gunnar, Madsen and Long2008).
Besides the peripheral effects such as stimulation of central adiposity, the concern is that both cortisol and pro-inflammatory cytokines cross the blood–brain barrier and can affect early brain development. Thus, Insel and others have proposed that the DOHaD principle applies to mental as well as physical health (Bale et al., Reference Bale, Baram, Brown, Goldstein, Insel, McCarthy and Nestler2010). Thus, while “fetal programming” remains an attractive and relatively easily studied construct, the definition of early life environmental effects on long-term neurodevelopment must be broadened to account for carryover effects from fetal life and the possibility that postnatal environmental events particularly during the first year affect life span neurodevelopment through similar mechanisms ascribed to “fetal programming.” Randomized clinical trials support this construct. Iron supplementation of pregnant women from early pregnancy to 12 weeks postpartum significantly increases performance on working memory and inhibitory control in their offspring at 7–9 years of age compared to children whose mothers were not supplemented (Christian et al., Reference Christian, Murray-Kolb, Khatry, Katz, Schaefer, Cole and Tielsch2010). In contrast, iron supplementation of 12- to 36-month-old children, whose mothers did not receive prenatal iron supplementation, had no effect on neurodevelopment at 7–9 years (Christian et al., Reference Christian, Morgan, Murray-Kolb, LeClerq, Khatry, Schaefer and Tielsch2011; Pongcharoen et al., Reference Pongcharoen, DiGirolamo, Ramakrishnan, Winichagoon, Flores and Martorell2011). Similarly, linear growth in the first 12 months postnatally strongly correlates with IQ at 9 years of age, but growth after 12 months had no impact (Pongcharoen et al., Reference Pongcharoen, Ramakrishnan, DiGirolamo, Winichagoon, Flores, Singkhornard and Martorell2012). Clearly, the fetal period through 12 months postnatal age is of particular importance in maintaining fetal and early postnatal nutritional homeostasis in order to optimize long-term development (Pollitt, Gorman, Engle, Martorell, & Rivera, Reference Pollitt, Gorman, Engle, Martorell and Rivera1993).
The duration of a term pregnancy in the human is 9 months and is typically divided into trimesters. Neurodevelopment can be affected by events in any of the fetal trimesters. Typically, events that occur in the first trimester have a larger impact on outcome than events in the third trimester because the neurologic processes that are disrupted are more fundamental in nature. These processes include structural embryogenesis in the first 8 weeks of pregnancy (when many mothers do not definitively know that they are pregnant), neurogenesis, and cell migration. The respective clinical syndromes of neural tube defect (e.g., spina bifida, anencephaly, and holoprosencephaly), microcephaly, and migration defects (e.g., lissencephaly) cause severe neurodevelopmental impairments. Events that occur in the third trimester typically cause less major disruptions but nevertheless can have significant impact on neural circuit construction. The latter is particularly important when relating fetal events to later risk of developmental psychopathology. All of the adverse fetal brain exposures discussed in this article follow these general rules of early versus late timing. For example, fetal alcohol exposure in the first trimester results in severe microcephaly and structural abnormalities, while the same exposure in the third trimester often spares head growth but results in more subtle deficits that may not become obvious for years (Bale et al., Reference Bale, Baram, Brown, Goldstein, Insel, McCarthy and Nestler2010).
A series of fundamental questions are thus raised when assessing environmental events in fetal life, brain development, and subsequent risk of psychopathology. Does altered early brain development due to fetal environmental events result in altered child and adult brain function, as indexed by altered behavior? If so, how predictable is the linkage? What accounts for the variability in behavioral outcomes following fetal environmental events? What are the mechanisms by which early life events permanently alter neural circuitry such that the behavioral phenotype remains abnormal despite correction of the abnormal fetal environment? Do disparate events that occur during a specific epoch in pregnancy result in a similar behavioral phenotype (i.e., unifinality) or does the nature of the fetal event matter in terms of structures that are affected?
To answer these questions, it is useful to consider the types of fetal environmental events that affect early brain development. Prenatal and early postnatal events affect early brain development by two basic pathways: events of omission and events of commission. Events of omission occur when factors important for normal brain development are absent. For example, premature delivery shortchanges the fetus of expectant intrauterine nutrients that are critical for brain development and are transported from mother to fetus during the third trimester. Not only does the prematurely born fetus fail to accrete those nutrients, but most preterm infants fail to thrive for months postnatally (Ehrenkranz, Reference Ehrenkranz2000) because they are difficult to nourish and because their metabolic needs are quite different than the in utero fetus.
Events of commission are defined as adverse events that happen to the brain because substances that should not be in the young brain are present. Classic examples of events of commission include fetal alcohol or teratogen exposure because these disruptors alter the architecture and metabolism of the developing brain. Clinically, the outcomes of many populations are a function of a combination of omitted and committed events. Events of commission can lead to events of omission. For example, fetal alcohol exposure, a clear event of commission, also reduces iron accretion by the fetal brain, leading to brain iron deficiency, a clear event of omission. Events of omission can also lead to secondary omission events. IUGR due to maternal malnutrition or maternal hypertension shortchanges the developing fetal brain of multiple nutrients that are critical for growth and development, including glucose, fatty acids, amino acids, iron, zinc, copper, folate, choline, and vitamin B12 (Rao & Georgieff, Reference Rao and Georgieff2002). However, infants with IUGR are also more vulnerable to neonatal encephalopathy at birth because of the fragile nature of their physiology. Neonatal encephalopathy is characterized by a reduced oxygen delivery to the brain, which is clearly an event of omission. The compounding of adverse events is frequently a greater risk to the brain than the individual components added together.
Behavior is the efferent expression of the brain (Hebb, Reference Hebb1949). Since Hebb's time, it has become apparent that while neuronal activity is the main driver of behavior, the glia play an important part in behavior as well. In addition, while neuronal anatomic complexity is closely correlated to functionality in certain brain structures (e.g., hippocampus), nonanatomic functions such as neuronal metabolism and neurotransmitter function are drivers of the brain's electrical potential. Environmental factors (e.g., fetal nutrition) can affect all cellular aspects of brain function from anatomy to electrophysiology. For example, fetal nutrients such as protein, fats, iron, zinc, and choline affect aspects of neuronal anatomy including cell number (cell division), size, and complexity. These same nutrients affect anatomical characteristics of oligodendrocytes that generate myelin and improve speed of processing, astrocytes that deliver nutrients and carry out reparative processes and microglia that are involved in cell trafficking and inflammatory responses. Astrocytes also provide both the “gliogenic” and “neurogenic niches,” which are likely important for stress responses in hippocampal neurogenesis, and cortical thickness (Horner & Palmer, Reference Horner and Palmer2003). The anatomic brain, however, is inert unless activated through its neurochemistry (e.g., neurotransmitters and ion channels) and its ability to generate action potentials (e.g., neurometabolism). The same nutrients responsible for anatomic integrity in the fetus also directly regulate neurotransmitter and neurotransmitter receptor expression as well as reuptake mechanisms. The fetal brain is the most metabolically active organ during development, consuming 60% of the body's total energy expenditure (Kuzawa, Reference Kuzawa1998). Much of that fetal energy metabolism is devoted to structural development including neurite extension and synaptogenesis during construction of neural circuits that underlie simple and complex behaviors and the energy required to support electrical activity of those circuits. Glucose, protein, iron, zinc, and choline have profound effects on mitochondrial and glycolytic metabolism that provide ATP to fuel that fetal growth and brain activity.
As noted earlier, the cost to society of early life environmental disruptors is through their long-term effects. Two major theories that are not mutually exclusive can account for long-term loss of synaptic efficacy. The first revolves around the concept of critical and sensitive periods and the effects of environmental events that occur during a critical or sensitive period. In this conceptual framework, adverse events that occur throughout a critical period of development result in permanent structural damage to the nervous system (Hensch, Reference Hensch2004). The impact of an adverse early life event on the adult behavioral phenotype will vary since each brain region and process is on a unique developmental trajectory. The permanence of the negative effect appears to depend on the adverse event lasting throughout the critical period. For example, neuronal iron deficiency disrupts hippocampal structure if it occurs during the period of rapid growth and differentiation (Postnatal Days 10 to 28 in the mouse). Repletion of iron within the critical period (i.e., Day 21) restores structural integrity and preserves adult memory function. In contrast, repletion outside of the critical window (i.e., Day 42) results in permanent structural and behavioral abnormalities (Callahan, Thibert, Wobken, & Georgieff, Reference Callahan, Thibert, Wobken and Georgieff2013; Carlson et al., Reference Carlson, Tkac, Magid, O'Connor, Andrews, Schallert and Petryk2009; Fretham et al., Reference Fretham, Carlson, Wobken, Tran, Petryk and Georgieff2012). In this conceptualization, the neurobehavioral deficits relate directly to the disordered neuronal structure and include poorer memory and stimulus gating (Fretham et al., Reference Fretham, Carlson, Wobken, Tran, Petryk and Georgieff2012; Jorgenson, Sun, O'Connor, & Georgieff, Reference Jorgenson, Sun, O'Connor and Georgieff2005; Pisansky et al., Reference Pisansky, Wickham, Su, Fretham, Yuan, Sun and Georgieff2013). The former is consistent with studies of early iron deficiency causing poorer neurocognitive function and an increased risk of schizophrenia in adulthood in humans (Insel, Schaefer, McKeague, Susser, & Brown, Reference Insel, Schaefer, McKeague, Susser and Brown2008; Lukowski et al., Reference Lukowski, Koss, Burden, Jonides, Nelson, Kaciroti and Lozoff2010).
The second conceptual framework revolves around the ability of early life events, particularly stress and specific nutritional deficiencies, to alter regulation of synaptic plasticity genes through epigenetic modifications (see below). Epigenetic modifications can involve methylation or hydroxyl-methylation of DNA and methylation, acetylation, and other biochemical changes of histones. The sum positive or negative effect of epigenetic modification typically cannot be judged by the degree of modification, but instead through understanding whether the genes that are modified are repressed or activated and whether the function of the genes are positive or negative.
Epigenetic modification is the most investigated biological mechanism underlying fetal programming to date. Fetal programming refers to the mechanisms by which early life environmental stimuli alter how genes are expressed throughout the lifetime. Epigenetics is the study of how supragenomic changes established early in life can permanently alter gene expression and resultant phenotype, independent of alterations to the genome itself. Mechanisms of epigenetic modification include DNA methylation, histone modification, and modulation by small noncoding RNAs, all of which are capable of altering patterns of gene expression. DNA methylation is particularly well characterized and includes two waves in which the genome is demethylated and then remethylated de novo, allowing for a complete “reprogramming” of the methylome. The second wave of remethylation establishes the methylation pattern that persists into mature somatic tissues and takes place during early embryogenesis. At this stage, DNA methylation is particularly vulnerable to intrauterine environmental exposures such as stress, toxicants, and malnutrition. Altered methylation of genes or their regulatory domains during this period can lead to lasting changes in gene expression and respective adult phenotypes (Bale et al., Reference Bale, Baram, Brown, Goldstein, Insel, McCarthy and Nestler2010; Heijmans, Tobi, Lumey, & Slagboom, Reference Heijmans, Tobi, Lumey and Slagboom2009; Simmons, Reference Simmons2005; Tobi et al., Reference Tobi, Lumey, Talens, Kremer, Putter, Stein and Heijmans2009).
Maternal/fetal stress clearly alters DNA methylation of brain-derived neurotrophic factor (BDNF), an important growth and differentiation growth factor for the brain (Lubin, Roth, & Sweatt, Reference Lubin, Roth and Sweatt2008). Growth factors are important for brain growth because they promote the effect of nutrients through signaling systems such as the mammalian target of rapamycin pathway. Neuronal mammalian target of rapamycin pathway activity is regulated by the presence of nutrients that are particularly important for growth and differentiation such as energy, protein, iron, oxygen, and zinc. Growth factors regulate the utilization of these essential nutrients to regulate actin polymerization as well as DNA transcription and protein translation rates, all of which in turn determine cell size and complexity (Fretham, Carlson, & Georgieff, Reference Fretham, Carlson and Georgieff2011; Wullschleger, Loewith, & Hall, Reference Wullschleger, Loewith and Hall2006). Reduction of BDNF levels in the hippocampus results in reduced downstream expression of genes that are critical for neuronal function. Lower BDNF levels during development are associated with abnormal neuronal structure in adulthood in preclinical models (Brunette, Tran, Wobken, Carlson, & Georgieff, Reference Brunette, Tran, Wobken, Carlson and Georgieff2010; Fretham et al., Reference Fretham, Carlson, Wobken, Tran, Petryk and Georgieff2012; Jorgenson et al., Reference Jorgenson, Sun, O'Connor and Georgieff2005).
Several fetal/neonatal nutritional conditions alter brain epigenetic status in preclinical models. IUGR, a model of generalized fetal malnutrition, disrupts hippocampal histone methylation (Ke et al., Reference Ke, Xing, Yu, Yu, Majnik, Cohen and Joss-Moore2014). It also alters BDNF DNA methylation (Ke et al., Reference Ke, Schober, McKnight, O'Grady, Caprau, Yu and Lane2010), potentially through inducing activation of the stress axis hormones. Long-chain polyunsaturated fatty acids (e.g., docosahexaenoic acid) found in fish oils also epigenetically modify BDNF through DNA methylation. Early life iron deficiency has little effect on BDNF methylation, but instead exerts its effects on the genome through a direct effect on a family of iron-containing histone demethylases (JARIDs/KDMs) that regulate BDNF expression (Blegen et al., Reference Blegen, Kennedy, Thibert, Gewirtz, Tran and Georgieff2013; Tran, Kennedy, Lien, Simmons, & Georgieff, Reference Tran, Kennedy, Lien, Simmons and Georgieff2015). Finally, certain nutrients in the one-carbon family (e.g., choline, folate, and betaine) can act as methyl donors and affect both DNA and histone methylation status (Barua et al., Reference Barua, Kuizon, Chadman, Flory, Brown and Junaid2014; Cho et al., Reference Cho, Sanchez-Hernandez, Reza-Lopez, Huot, Kim and Anderson2013; Zeisel, Reference Zeisel2017).
In summary, fetal environmental events can permanently alter the expression of genes that are critical to adult neural function. The effects can be circuit and region specific, thereby potentially altering the activity of circuits and leading to aberrant behavioral phenotypes in adulthood. The critical period and epigenetic conceptual frameworks are not mutually exclusive as altered structure will result in altered gene expression and vice versa.
For each of the fetal clinical conditions considered in the following sections, we explore the evidence at the epidemiologic, clinical, preclinical, and molecular levels that supports a causative association between the fetal condition and later development of psychopathology. The range of conditions represents both events of commission (fetal alcohol syndrome and teratogen exposure) and events of commission (single or multiple nutrient deficits). While it is tempting to ascribe a direct cause and effect pathway for any of these conditions, it is likely that any of the psychopathologies that are proposed to have developmental origins (including schizophrenia, autism spectrum disorder, depression/anxiety, and attention-deficit/hyperactivity disorder) are polygenic, and potentially polyenvironmental in their etiologies (Bale et al., Reference Bale, Baram, Brown, Goldstein, Insel, McCarthy and Nestler2010; Basavarajappa & Subbanna, Reference Basavarajappa and Subbanna2016). Careful dissection of the relative contribution of each early life factor may identify potential treatment and prevention targets in order to reduce the overall risk of disease appearance later in life.
Fetal Alcohol Spectrum Disorder (FASD) and Its Relationship to Developmental Psychopathology
Epidemiological and clinical studies
Prenatal alcohol exposure is considered the most common cause of mental retardation (National Institute on Alcohol Aabuse and Alcoholism, 1990). High rates of psychopathology are seen in children with FASD, including externalizing and internalizing behaviors, sleep disorders, abnormal habits and stereotypies, and behavioral management problems, even in the absence of intellectual disability (Steinhausen, Willms, & Spohr, Reference Steinhausen, Willms and Spohr1993; Streissguth et al., Reference Streissguth, Aase, Clarren, Randels, LaDue and Smith1991). The high rates of psychopathology persist in adulthood and include substance disorders, mood disorders, psychotic disorders, personality disorders, anxiety disorders, and eating disorders (Famy, Streissguth, & Unis, Reference Famy, Streissguth and Unis1998). Fetal alcohol damage is associated with later onset of schizophrenia (Slavney & Grau, Reference Slavney and Grau1978). Attention-deficit/hyperactivity disorder (ADHD) was found to be the most common comorbid disorder among children with FASD (52.9%), followed by oppositional defiant disorder (12.9%), conduct disorder (7.0%), and autism spectrum disorder (ASD; 2.6%; Lange, Rehm, Anagnostou, & Popova, Reference Lange, Rehm, Anagnostou and Popova2017). While FASD is associated with global executive impairments, executive function weaknesses are most commonly found and consist of impairments of planning, fluency, and set shifting (Kingdon, Cardoso, & McGrath, Reference Kingdon, Cardoso and McGrath2016).
FASD results in changes to multiple neural circuits that can be imaged noninvasively in humans. High incidences of midline brain abnormalities of the corpus callosum such as hypoplasia or agenesis are seen after prenatal alcohol exposure (Bookstein et al., Reference Bookstein, Connor, Covell, Barr, Gleason, Sze and Streissguth2005, Reference Bookstein, Connor, Huggins, Barr, Pimentel and Streissguth2007; Bookstein, Sampson, Streissguth, & Connor, Reference Bookstein, Sampson, Streissguth and Connor2001; Riley et al., Reference Riley, Mattson, Sowell, Jernigan, Sobel and Jones1995; Sowell, Mattson, et al., Reference Sowell, Mattson, Thompson, Jernigan, Riley and Toga2001; Sowell, Thompson, et al., Reference Sowell, Thompson, Mattson, Tessner, Jernigan, Riley and Toga2001; Swayze et al., Reference Swayze, Johnson, Hanson, Piven, Sato, Giedd and Andreasen1997). More recent studies examining white matter microstructure with diffusion tensor imaging have found global white matter changes that could account for dysfunction of multiple behavioral domains including working memory, mathematical ability, executive function, processing speed, intelligence, and interhemispheric transfer (Donald, Eastman, et al., Reference Donald, Eastman, Howells, Adnams, Riley, Woods and Stein2015; Donald, Roos, et al., Reference Donald, Roos, Fouche, Koen, Howells, Woods and Stein2015; Fan et al., Reference Fan, Jacobson, Taylor, Molteno, Dodge, Stanton and Meintjes2016; Fryer et al., Reference Fryer, Schweinsburg, Bjorkquist, Frank, Mattson, Spadoni and Riley2009; Lebel, Rasmussen, Wyper, Andrew, & Beaulieu, Reference Lebel, Rasmussen, Wyper, Andrew and Beaulieu2010; Lebel et al., Reference Lebel, Rasmussen, Wyper, Walker, Andrew, Yager and Beaulieu2008; Malisza et al., Reference Malisza, Buss, Bolster, de Gervai, Woods-Frohlich, Summers and Longstaffe2012; Spottiswoode et al., Reference Spottiswoode, Meintjes, Anderson, Molteno, Stanton, Dodge and Jacobson2011; Taylor et al., Reference Taylor, Jacobson, van der Kouwe, Molteno, Chen, Wintermark and Meintjes2015; Wozniak et al., Reference Wozniak, Mueller, Chang, Muetzel, Caros and Lim2006, Reference Wozniak, Muetzel, Mueller, McGee, Freerks, Ward and Lim2009). FASD is also associated with changes in functional connectivity in cortical networks (medial prefrontal and posterior cingulate), the default mode network, corticostriatal networks (affecting working memory), and whole-brain connectivity (associated with lower global cognitive functioning; Roussotte et al., Reference Roussotte, Sulik, Mattson, Riley, Jones, Adnams and Sowell2012; Santhanam et al., Reference Santhanam, Coles, Li, Li, Lynch and Hu2011; Wozniak et al., Reference Wozniak, Mueller, Muetzel, Bell, Hoecker, Nelson and Lim2011, Reference Wozniak, Mueller, Mattson, Coles, Kable, Jones and Sowell2017). Hippocampal volume and declarative memory deficits are prominent long-term features of FASD in humans (Autti-Ramo et al., Reference Autti-Ramo, Autti, Korkman, Kettunen, Salonen and Valanne2002; Willoughby, Sheard, Nash, & Rovet, Reference Willoughby, Sheard, Nash and Rovet2008). The degree of hippocampal volume loss in humans is directly correlated with the amount of memory deficit (Autti-Ramo et al., Reference Autti-Ramo, Autti, Korkman, Kettunen, Salonen and Valanne2002; Willoughby et al., Reference Willoughby, Sheard, Nash and Rovet2008).
Preclinical models and molecular mechanisms
Preclinical models have investigated the effect of fetal alcohol exposure on the developing brain and the subsequent consequences to the adult brain. The behavioral findings in the preclinical model animals, including rats, mice, and nonhuman primates, corroborate the findings in humans. Analysis of the brains from the models has provided biological plausibility at the regional anatomic and circuit level to support the hypothesis that fetal alcohol exposure results in psychopathology at an older age in humans. Molecular analyses of the brains from the models provide insights into the mechanisms of the long-term findings. In general, the long-term effects of fetal alcohol exposure follow the temporal and regionalization rules seen with nutrient deficiencies (Kretchmer, Beard, & Carlson, Reference Kretchmer, Beard and Carlson1996; Maier, Miller, Blackwell, & West, Reference Maier, Miller, Blackwell and West1999). Alcohol exposure during different trimesters in the rat resulted in regional brain area differences of cell loss and also contrasted with alcohol exposure throughout the entire pregnancy (Maier et al., Reference Maier, Miller, Blackwell and West1999).
Preclinical models demonstrate that alcohol exposure results in both global and regional specific effects. Global effects are particularly prominent with early to middle gestation onset of exposure. Two doses of alcohol on Day 9 of a 21-day gestation results in volume and significant misshaping of the cerebral cortex, hippocampus, striatum, and cerebellum (Parnell et al., Reference Parnell, Holloway, O'Leary-Moore, Dehart, Paniaqua, Oguz and Sulik2013). The regional effects of fetal alcohol exposure in the rodent demonstrate that the hippocampus is particularly vulnerable (Berman & Hannigan, Reference Berman and Hannigan2000; Livy, Miller, Maier, & West, Reference Livy, Miller, Maier and West2003; Miller, Reference Miller1995), supporting similar findings in humans (Autti-Ramo et al., Reference Autti-Ramo, Autti, Korkman, Kettunen, Salonen and Valanne2002; Willoughby et al., Reference Willoughby, Sheard, Nash and Rovet2008). All subregions of the hippocampus are affected in rodent models. The dentate gyrus and CA1 have reduced neuronal number (Klintsova et al., Reference Klintsova, Helfer, Calizo, Dong, Goodlett and Greenough2007; Miller, Reference Miller1995). Exposure restricted to the third trimester reduces the volume of CA1 due to lower cell number and reduced pyramidal cell density (Livy et al., Reference Livy, Miller, Maier and West2003). Prenatal alcohol exposure also damages area CA3 (Ba, Seri, Aka, Glin, & Tako, Reference Ba, Seri, Aka, Glin and Tako1999).
The loss of hippocampal anatomy is accompanied by loss of synaptic plasticity across the life span in rodent models (Hablitz, Reference Hablitz1986; Krahl, Berman, & Hannigan, Reference Krahl, Berman and Hannigan1999; Swartzwelder, Farr, Wilson, & Savage, Reference Swartzwelder, Farr, Wilson and Savage1988; Tan, Berman, Abel, & Zajac, Reference Tan, Berman, Abel and Zajac1990). In vitro electrophysiological studies in hippocampal brain slices isolated from adult animals following prenatal alcohol-exposed animals demonstrate changes in synaptic activity (Berman & Hannigan, Reference Berman and Hannigan2000). The loss of electrophysiologic potential may result from abnormalities in glutamatergic neurotransmission (Kimura, Reynolds, & Brien, Reference Kimura, Reynolds and Brien2000; Reynolds & Brien, Reference Reynolds and Brien1995; Thomas, Fleming, & Riley, Reference Thomas, Fleming and Riley2001). Fetal alcohol exposure also results in brain iron deficiency (Helfrich, Saini, Kling, & Smith, Reference Helfrich, Saini, Kling and Smith2017; Huebner, Blohowiak, Kling, & Smith, Reference Huebner, Blohowiak, Kling and Smith2016; Rufer et al., Reference Rufer, Tran, Attridge, Andrzejewski, Flentke and Smith2012), which in turn compromises glutamatergic neurotransmission (Jorgenson et al., Reference Jorgenson, Sun, O'Connor and Georgieff2005; Pisansky et al., Reference Pisansky, Wickham, Su, Fretham, Yuan, Sun and Georgieff2013; Rao, Tkac, Townsend, Gruetter, & Georgieff, Reference Rao, Tkac, Townsend, Gruetter and Georgieff2003).
The translation of alcohol-induced hippocampal deficits from preclinical models to human long-term behavioral abnormalities extends beyond declarative memory deficits because the hippocampus is also central in circuits that support more complex behaviors. For example, tonic hippocampal input is critical for regulation of the VTA loop. Disruption of this input through compromise of hippocampal function with resulting imbalance between hippocampal and prefrontal cortical inputs has been proposed as a mechanism in schizophrenia (Esmaeili & Grace, Reference Esmaeili and Grace2013; White, Joseph, Francis, & Liddle, Reference White, Joseph, Francis and Liddle2010). Dysregulation of the VTA loop has also been implicated in drug addiction research, and as noted above, the rate of drug addiction is higher in adults with FASD (Macht, Kelly, & Gass, Reference Macht, Kelly and Gass2017). The hippocampus is central to the circuitry involved in sensory gating, a key behavior that is disordered in schizophrenia (Pisansky et al., Reference Pisansky, Wickham, Su, Fretham, Yuan, Sun and Georgieff2013). Finally, the integrity of the hippocampus in early life is important in providing scaffolding for frontal lobe development later in childhood. While the prefrontal cortex may be directly injured by fetal alcohol exposure, it may also receive poor connections from an injured limbic system (Wachs, Georgieff, Cusick, & McEwen, Reference Wachs, Georgieff, Cusick and McEwen2014). Fetal insults that initially cause declarative learning and memory deficits in the newborn period can manifest as attentional issues at older ages (Deregnier, Nelson, Thomas, Wewerka, & Georgieff, Reference Deregnier, Nelson, Thomas, Wewerka and Georgieff2000; Jabes, Thomas, Langworthy, Georgieff, & Nelson, Reference Jabes, Thomas, Langworthy, Georgieff and Nelson2015).
In humans, prenatal alcohol exposure modifies the somatic cell DNA methylation (Portales-Casamar et al., Reference Portales-Casamar, Lussier, Jones, MacIsaac, Edgar, Mah and Kobor2016), providing evidence for potential epigenetic mechanism underlying the long-term changes in gene expression. Certainly, more studies are needed to establish this relationship. In animal models, fetal alcohol exposure alters expression of neural genes critical for important cellular processes, including proliferation, fate specification, migration, and differentiation (Kleiber, Mantha, Stringer, & Singh, Reference Kleiber, Mantha, Stringer and Singh2013; Sanchez-Alvarez, Gayen, Vadigepalli, & Anni, Reference Sanchez-Alvarez, Gayen, Vadigepalli and Anni2013). These findings provide a molecular and cellular basis to explicate the neurodevelopmental deficits associated with prenatal alcohol exposure. The relationship between prenatal alcohol exposure and epigenetic modifications is more evident in the animal models. In line with limited data from humans, prenatal alcohol modifies DNA methylation patterns in mouse embryos (Hicks, Middleton, & Miller, Reference Hicks, Middleton and Miller2010; Liu, Balaraman, Wang, Nephew, & Zhou, Reference Liu, Balaraman, Wang, Nephew and Zhou2009), expression of noncoding RNAs implicated in epigenetic modifications (Laufer et al., Reference Laufer, Mantha, Kleiber, Diehl, Addison and Singh2013), and neural expression of DNA methylation modifiers such as DNMTs and MeCP2 (Gangisetty, Bekdash, Maglakelidze, & Sarkar, Reference Gangisetty, Bekdash, Maglakelidze and Sarkar2014; Kim et al., Reference Kim, Park, Choi, Choi, Joo, Kim and Shin2013; Liyanage, Zachariah, Davie, & Rastegar, Reference Liyanage, Zachariah, Davie and Rastegar2015; Perkins, Lehmann, Lawrence, & Kelly, Reference Perkins, Lehmann, Lawrence and Kelly2013). Emerging evidence has also implicated prenatal alcohol exposure in modifying histone acetylation and methylation as another epigenetic link to the long-term gene dysregulation (Chater-Diehl, Laufer, & Singh, Reference Chater-Diehl, Laufer and Singh2017).
Preclinical Models of Teratogen Exposure (Bisphenol-A) and Their Relation to Developmental Psychopathology
Teratogens represent a major “commission” risk to the developing fetal brain. Teratogens typically fall into the categories of drug exposures and environmental toxicants. Alcohol (discussed above) easily fits into the teratogen category as an abnormal drug exposure. In all cases, they represent environmental molecules that are not needed for normal brain development. It is beyond the scope of this article to catalog the suspected brain effects of all teratogens. The following discussion focuses on bisphenol-A (BPA) because of growing evidence that potential endocrine disruptors during pregnancy significantly affect brain development.
Epidemiological and clinical studies
Fetal-neonatal exposure to BPA, a chemical used in the production of commonly used plastics, is associated with dysfunction in metabolism and reproductive development (Dolinoy, Huang, & Jirtle, Reference Dolinoy, Huang and Jirtle2007). BPA can traverse the placenta and is found in fetal serum and full-term amniotic fluid, with highest levels found between 15 and 18 weeks gestation (Ikezuki, Tsutsumi, Takai, Kamei, & Taketani, Reference Ikezuki, Tsutsumi, Takai, Kamei and Taketani2002). In humans, while the long-term effects of early life BPA exposure on neurobehavioral development remain controversial (Casas et al., Reference Casas, Forns, Martinez, Avella-Garcia, Valvi, Ballesteros-Gomez and Vrijheid2015), accumulating evidence suggests a positive association between BPA exposure and risk of ADHD in male (Philippat et al., Reference Philippat, Nakiwala, Calafat, Botton, De Agostini, Heude and Slama2017; Tewar et al., Reference Tewar, Auinger, Braun, Lanphear, Yolton, Epstein and Froehlich2016), internalizing and externalizing behavior in male (Evans et al., Reference Evans, Kobrosly, Barrett, Thurston, Calafat, Weiss and Swan2014), and social communication deficit in female children (Lim et al., Reference Lim, Bae, Kim, Shin, Lee, Kim and Hong2017; Miodovnik et al., Reference Miodovnik, Engel, Zhu, Ye, Soorya, Silva and Wolff2011). Braun et al. (Reference Braun, Yolton, Dietrich, Hornung, Ye, Calafat and Lanphear2009) found a relationship between prenatal BPA exposure and externalizing behavior in 2-year-old girls but not boys using the Behavioral Assessment System for Children. Gestational urinary BPA concentrations were associated with some neurobehavioral measures at 3 years of age in this cohort (prospective birth cohort, 244 mothers). In particular, gestational BPA exposure was associated with higher scores for measures of anxiety, hyperactivity, emotional control, and behavioral inhibition (internalizing and externalizing behaviors). Results were more pronounced for externalizing behaviors: girls exhibited increases in hyperactivity, and boys exhibited decreases in hyperactivity (Braun & Hauser, Reference Braun and Hauser2011; Tewar et al., Reference Tewar, Auinger, Braun, Lanphear, Yolton, Epstein and Froehlich2016). Conversely, another study found that among boys (but not girls), prenatal BPA exposure was significantly associated with emotionally reactive and aggressive behaviors and higher syndrome scores (internalizing and externalizing problems) between the ages of 3 and 5 (Perera et al., Reference Perera, Vishnevetsky, Herbstman, Calafat, Xiong, Rauh and Wang2012). Another study found no correlation between maternal BPA and infant neurobehavioral abnormalities in 5-week-old infant neurobehaviors; however, the authors noted that BPA concentrations in the maternal samples may have been below the threshold of neurobehavioral effects (Yolton et al., Reference Yolton, Xu, Strauss, Altaye, Calafat and Khoury2011).
Preclinical models and molecular mechanisms
The findings in humans suggest a connection between BPA and brain circuits that mediate ADHD, schizophrenia, social communication deficits, and internalizing/externalizing disorders. The biology underlying the behavioral deficits observed in human studies derives from the bodies of work in animal models and implicates abnormal structure in the hippocampus, caudate nucleus, and prefrontal cortex as well as significant effects on at least two neurotransmitter systems. These cellular and structural changes likely underlie the behavioral effects of prenatal BPA exposure.
BPA, like nutrient deficiencies, follows the rules of timing and regionalization. Rat dams were exposed to BPA in the last trimester of pregnancy and then randomized to additional exposure or placebo postnatally. Open field and elevated plus maze tests of anxiety-like behaviors administered during the juvenile and adult periods demonstrated that prenatal exposure reduced exploratory behavior and increased anxiety particularly in females (Gioiosa, Parmigiani, Vom Saal, & Palanza, Reference Gioiosa, Parmigiani, Vom Saal and Palanza2013). The sexually dimorphic behavior may be due to inhibition of estrogen receptor-beta (Xu et al., Reference Xu, Wang, Zhang, Luo, Ye and Ruan2010).
Fetal and neonatal BPA exposure in mice compromises recognition memory as evidenced by poorer performance on the novel object recognition test, a finding that suggests compromise of the developing hippocampus (Tian, Baek, Lee, & Jang, Reference Tian, Baek, Lee and Jang2010). Adverse effects are seen at both the neuroanatomical and the neurotransmitter receptor levels. Reduced recognition memory is accompanied by lower NMDA receptor, consistent with alterations to the glutamatergic neurotransmitter system that is dominant in that brain area. The NMDA receptor effect is dose dependent. Low-dose BPA exposure decreases NMDA concentration, whereas high doses more selectively suppress NR1 than NR2A or B. This is developmentally important because NR1 combines first with NR2A and then developmentally switches to NR2B when long-term potentiation matures. BPA-induced alteration of subunits expression would be expected to alter hippocampal electrophysiologic development and capacity (Jorgenson et al., Reference Jorgenson, Sun, O'Connor and Georgieff2005). The hippocampus of prenatally BPA exposed monkeys also contains fewer dopaminergic neurons (Elsworth et al., Reference Elsworth, Jentsch, Vandevoort, Roth, Redmond and Leranth2013), suggesting compromise of a second neurotransmitter system. While the hippocampus has relatively low amounts of dopamine, its presence is important for providing salience to fear memories. Dopamine blockade increases the rate of fear memory extinction, whereas dopamine agonists slow the extinction rate (Menezes et al., Reference Menezes, Alves, Borges, Roehrs, de Carvalho Myskiw, Furini and Mello-Carpes2015; Ponnusamy, Nissim, & Barad, Reference Ponnusamy, Nissim and Barad2005). The finding of fewer dopaminergic neurons is consistent with the reduced anxiety and fear that BPA-exposed mice demonstrate on open field and elevated plus maze tests (Tian et al., Reference Tian, Baek, Lee and Jang2010).
Prenatal BPA exposure disrupts at least three hormone systems that have a role in brain development. In addition to the effect on estrogen receptor beta (Xu et al., Reference Xu, Wang, Zhang, Luo, Ye and Ruan2010), stress-axis hormones may also be involved as fetal/neonatal BPA exposure alters glucocorticoid receptors more in females than males accompanied by greater anxiety in female offspring in adulthood (Poimenova, Markaki, Rahiotis, & Kitraki, Reference Poimenova, Markaki, Rahiotis and Kitraki2010). Furthermore, fetal/neonatal exposure acts as a thyroid hormone receptor antagonist in rats, potentially resetting neuronal metabolic set points and mitochondrial activity (Zoeller, Bansal, & Parris, Reference Zoeller, Bansal and Parris2005). Reduction of neuronal metabolic activity would result in simpler dendritic structure and consequent loss of function in the same manner as seen with nutrient deficiencies (Bastian, von Hohenberg, Mickelson, Lanier, & Georgieff, Reference Bastian, von Hohenberg, Mickelson, Lanier and Georgieff2016).
The brain regions and neurotransmitter systems that are altered permanently by fetal/neonatal BPA exposure reside in neural circuits that mediate important complex behaviors including the VTA loop and mesolimbic–prefrontal connections. The involvement of these circuits’ anatomy and neurochemistry provides a plausible biological basis for the psychopathology reported in human juveniles and adults, including anxiety, internalizing/externalizing behaviors, and schizophrenia.
In mice, there are evidence that fetal-neonatal BPA exposure alters cortical development by accelerating neuronal differentiation and migration during the early embryonic stage, resulting in a mistiming of critical development that had lasting effects into adulthood. The alterations were accompanied by changes to promoter-associated CpG islands suggesting that the long-term effects were epigenetic in nature (Itoh, Yaoi, & Fushiki, Reference Itoh, Yaoi and Fushiki2012) Whole-genome DNA methylation is altered in a dose-dependent manner by BPA administered in physiologically relevant concentrations during pregnancy to the agouti mouse (Anderson et al., Reference Anderson, Nahar, Faulk, Jones, Liao, Kannan and Dolinoy2012). Provision of a diet enhanced with methyl donors (e.g., folate) that are known to modify CpG island and histone methylation status counteracts the long-term epigenetic effects of fetal/neonatal PBA exposure (Dolinoy et al., Reference Dolinoy, Huang and Jirtle2007).
Similarly to the epigenetic findings with early life BPA exposure, three recent studies showed that developmental exposure to Atrazine, a commonly used agricultural herbicide, also induces changes in histone methylation and DNA methylation patterns in the rodent germ cells across multiple generations, thereby linking Atrazine exposure to stable and transmissible epigenetic modifications (Gely-Pernot et al., Reference Gely-Pernot, Hao, Becker, Stuparevic, Kervarrec, Chalmel and Smagulova2015; Hao et al., Reference Hao, Gely-Pernot, Kervarrec, Boudjema, Becker, Khil and Smagulova2016; McBirney et al., Reference McBirney, King, Pappalardo, Houser, Unkefer, Nilsson and Skinner2017). The effects on brain development remain to be determined.
Preclinical Models of Select Nutrient Deficiencies and Their Relation to Developmental Psychopathology
There is a substantial body of literature on the effect of fetal nutrition on adult mental health. All nutrients are important for brain development during the last trimester, but certain nutrients have a greater acute and lasting impact on the brain and behavior. These include macronutrients such as glucose, protein, and fats (especially long-chain polyunsaturated fatty acids) and micronutrients such as iron, zinc, copper, iodine, folate, choline, and vitamin B12. These higher impact nutrients demonstrate critical or sensitive periods for neurodevelopment where early life deficiency results in long-term dysfunction (Table 1). Several, including long-chain polyunsaturated fatty acids, iron, folate, and choline, as well as generalized fetal malnutrition, confer these long-term risks through epigenetic mechanisms (Georgieff, Brunette, & Tran, Reference Georgieff, Brunette and Tran2015; Grissom & Reyes, Reference Grissom and Reyes2013; Ke et al., Reference Ke, Xing, Yu, Yu, Majnik, Cohen and Joss-Moore2014; Tran et al., Reference Tran, Kennedy, Lien, Simmons and Georgieff2015; Tyagi, Zhuang, Agrawal, Ying, & Gomez-Pinilla, Reference Tyagi, Zhuang, Agrawal, Ying and Gomez-Pinilla2015; Zeisel, Reference Zeisel2017). A comprehensive review of each nutrient is beyond the scope of this article, but the principles are evident from well-studied nutrients such as iron deficiency and generalized fetal malnutrition, also referred to as IUGR. Both have been associated with an increased incidence of later onset of significant developmental psychopathologies such as schizophrenia (Eide et al., Reference Eide, Moster, Irgens, Reichborn-Kjennerud, Stoltenberg, Skjaerven and Abel2013; Insel et al., Reference Insel, Schaefer, McKeague, Susser and Brown2008), autism (Schmidt, Tancredi, Krakowiak, Hansen, & Ozonoff, Reference Schmidt, Tancredi, Krakowiak, Hansen and Ozonoff2014) and depression/anxiety (Lukowski et al., Reference Lukowski, Koss, Burden, Jonides, Nelson, Kaciroti and Lozoff2010) and thus merit in-depth analyses across all levels of investigation from epidemiology to molecular biology. They stand as valuable paradigms for the biological plausibility of early nutrition effects on adult mental health (Pollitt et al., Reference Pollitt, Gorman, Engle, Martorell and Rivera1993).
Table 1. Importance of timing of nutrient deficiency on long-term brain developmental outcomes
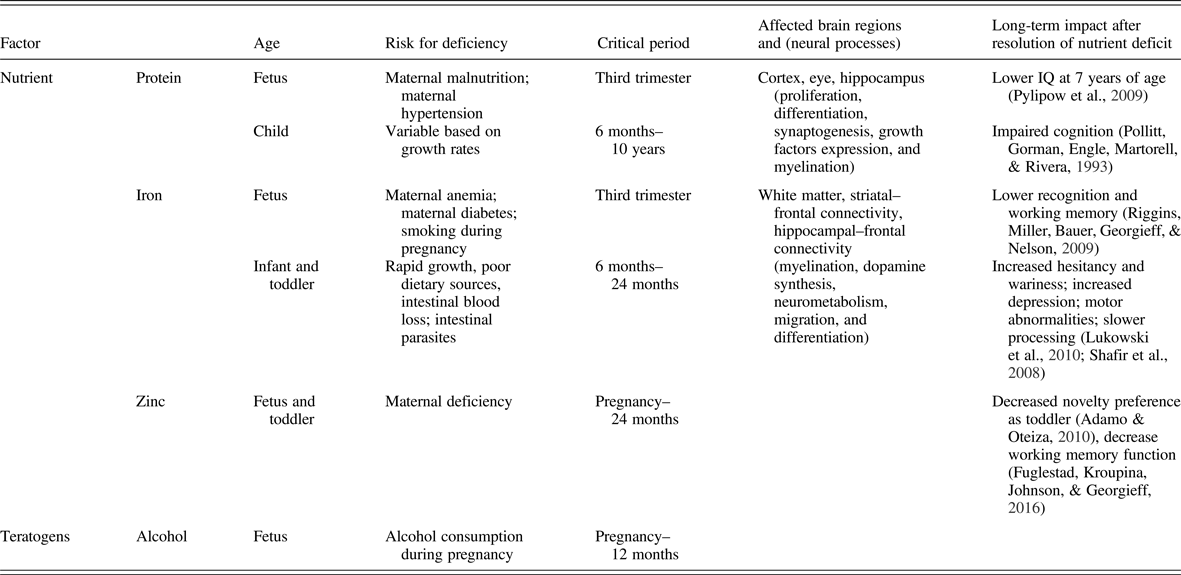
Epidemiological and clinical studies of iron deficiency
Iron deficiency (ID) is the most common nutrient deficiency in the world (Walker et al., Reference Walker, Wachs, Gardner, Lozoff, Wasserman, Pollitt and Carter2007). Fetal ID that affects brain development occurs in the context of maternal diabetes mellitus during pregnancy, maternal hypertension during pregnancy, chronic severe ID, or maternal anemia due to ID (Beard, Reference Beard2001; Lozoff et al., Reference Lozoff, Beard, Connor, Barbara, Georgieff and Schallert2006; Siddappa, Rao, Long, Widness, & Georgieff, Reference Siddappa, Rao, Long, Widness and Georgieff2007). Multiple studies have demonstrated the association between gestational diabetes in the mother and subsequent risk of developmental psychopathologies, particularly ASD and schizophrenia, in the offspring. Gestational diabetes mellitus is more common among mothers who have children with ASD and is associated with greater deficits in expressive language among children with ASD (Krakowiak et al., Reference Krakowiak, Walker, Bremer, Baker, Ozonoff, Hansen and Hertz-Picciotto2012). The risk for ASD is increased for children of women with diabetes (Leonard, de Klerk, Bourke, & Bower, Reference Leonard, de Klerk, Bourke and Bower2006). Preexisting type 2 diabetes mellitus and exposure to gestational diabetes diagnosed before (but not after) 26 weeks gestation is associated with an increased risk of ASD in offspring (Xiang et al., Reference Xiang, Wang, Martinez, Walthall, Curry, Page and Getahun2015). In a meta-analysis, diabetes during pregnancy was significantly associated with development of schizophrenia (Cannon, Jones, & Murray, Reference Cannon, Jones and Murray2002). Uncontrolled maternal diabetes also resulted in poorer performance on measures of intelligence, verbal ability, acquired knowledge, spatial ability, and sequence abilities (Rizzo, Metzger, Dooley, & Cho, Reference Rizzo, Metzger, Dooley and Cho1997). Gestational diabetes reduced expressive language performance in infants of diabetic mothers at 18, 30, and 72/84 months (Dionne, Boivin, Seguin, Perusse, & Tremblay, Reference Dionne, Boivin, Seguin, Perusse and Tremblay2008). An increased risk for mild to moderate intellectual disability was found in children of mothers with diabetes (Leonard et al., Reference Leonard, de Klerk, Bourke and Bower2006). The role of ID in linking maternal diabetes to poorer brain development was demonstrated in infants of diabetic mothers born with low cord ferritin concentrations indicating fetal ID. These infants have behavioral and electrophysiological measures of poorer recognition memory throughout the first 12 months of life in spite of resolution of their neonatal ID (DeBoer, Wewerka, Bauer, Georgieff, & Nelson, Reference DeBoer, Wewerka, Bauer, Georgieff and Nelson2005; Nelson, Wewerka, Borscheid, Deregnier, & Georgieff, Reference Nelson, Wewerka, Borscheid, Deregnier and Georgieff2003; Nelson et al., Reference Nelson, Wewerka, Thomas, Tribby-Walbridge, Regnier and Georgieff2000; Siddappa et al., Reference Siddappa, Georgieff, Wewerka, Worwa, Nelson and Deregnier2004).
The role of early life ID and anemia (most commonly due to ID) in psychopathology has been assessed independent of maternal diabetes during pregnancy. For mothers with anemia, there is increased risk of severe intellectual disability in their offspring (Hablitz, Reference Hablitz1986). Fetal/neonatal ID results in behavioral and electrophysiological measures of poor recognition memory of mother's voice at 2 months of age (Geng et al., Reference Geng, Mai, Zhan, Xu, Zhao, Georgieff and Lozoff2015). Early postnatal ID in Chilean infants is associated with attentional control deficits at age 10 years and heightened risk taking, excessive alcohol use, and risky sexual behavior in adolescence (East et al., Reference East, Lozoff, Blanco, Delker, Delva, Encina and Gahagan2017). Iron deficiency anemia (IDA) during early infancy is also associated with dull affect and social reticence at age 5, leading to functional isolation later in childhood (East et al., Reference East, Lozoff, Blanco, Delker, Delva, Encina and Gahagan2017). IDA results in long-lasting effects such as slower reaction time and poorer inhibitory control (with correlated EEG findings) on a Go/No-Go task in children 8 to 9 years after diagnosis of ID and treatment with iron supplementation (Algarin et al., Reference Algarin, Nelson, Peirano, Westerlund, Reyes and Lozoff2013). Infants with ID have adversely affected social–emotional behaviors such as increased shyness and latency to interact with an examiner, decreased positive affect and soothability (Lozoff et al., Reference Lozoff, Clark, Jing, Armony-Sivan, Angelilli and Jacobson2008), as well as poor object permanence and memory encoding and retrieval (Carter et al., Reference Carter, Jacobson, Burden, Armony-Sivan, Dodge, Angelilli and Jacobson2010). The effects of ID are long lasting, with decreased performance on measures of executive functioning and recognition memory at 19 years of age (Lukowski et al., Reference Lukowski, Koss, Burden, Jonides, Nelson, Kaciroti and Lozoff2010). This same group has now been followed out to 25 years, with formerly ID subjects having less completion of secondary school and fewer long-term relationships, and more negative emotions and feelings of detachment (Lozoff et al., Reference Lozoff, Smith, Kaciroti, Clark, Guevara and Jimenez2013). Case control studies have shown that infants with ID have lower scores on measures of mental and motor development, alterations in visual recognition memory, and improvement in these measures with iron supplementation (Friel et al., Reference Friel, Aziz, Andrews, Harding, Courage and Adams2003; Lozoff et al., Reference Lozoff, De Andraca, Castillo, Smith, Walter and Pino2003; Moffatt, Longstaffe, Besant, & Dureski, Reference Moffatt, Longstaffe, Besant and Dureski1994; Riggins, Miller, Bauer, Georgieff, & Nelson, Reference Riggins, Miller, Bauer, Georgieff and Nelson2009; Shafir, Angulo-Barroso, Calatroni, Jimenez, & Lozoff, Reference Shafir, Angulo-Barroso, Calatroni, Jimenez and Lozoff2006; Sherriff, Emond, Bell, & Golding, Reference Sherriff, Emond, Bell and Golding2001). Children with ID in infancy have poorer performance on tests of some specific cognitive functions such as executive function requiring inhibition and planning at 5 and 10 years, and spatial memory and selective attention in 11- to 14-year-olds (Congdon et al., Reference Congdon, Westerlund, Algarin, Peirano, Gregas, Lozoff and Nelson2012; Lozoff, Jimenez, Hagen, Mollen, & Wolf, Reference Lozoff, Jimenez, Hagen, Mollen and Wolf2000).
Recent neuroimaging studies are beginning to provide anatomic evidence of altered neonatal brain structure due to fetal ID. A diffusion tensor imaging study showed that low maternal prenatal iron status alters markers of normal development of axonal pathways (such as corpus callosum, internal capsule, and the longitudinal fasciculus) and frontal cortical gray matter (Monk et al., Reference Monk, Georgieff, Xu, Hao, Bansal, Gustafsson and Peterson2016). In a resting-state magnetic resonance imaging (MRI) study, former IDA subjects showed alterations in default mode and dorsal attention networks (Algarin et al., Reference Algarin, Karunakaran, Reyes, Morales, Lozoff, Peirano and Biswal2017).
Preclinical models and molecular mechanisms of fetal/neonatal ID
In animal models, fetal/neonatal ID showed similar neurological and behavioral deficits to the human cohorts (Lozoff & Georgieff, Reference Lozoff and Georgieff2006). Fetal/neonatal ID during critical periods of cortical development increases the risk of autism, ADHD, and schizophrenia pathogenesis (Erikson, Jones, & Beard, Reference Erikson, Jones and Beard2000; Gambling, Kennedy, & McArdle, Reference Gambling, Kennedy and McArdle2011; Schubert, Martens, & Kolk, Reference Schubert, Martens and Kolk2015). Preclinical models provide evidence for acute and long-term abnormalities in neuronal structure, connectivity, metabolism, and plasticity that are accompanied by changes in gene expression (Tran et al., Reference Tran, Kennedy, Pisansky, Won, Gewirtz, Simmons and Georgieff2016).
The neurobiological importance of iron in optimal fetal/neonatal brain development is well established (Lozoff & Georgieff, Reference Lozoff and Georgieff2006; Rao, Tkac, Schmidt, & Georgieff, Reference Rao, Tkac, Schmidt and Georgieff2011). Iron is a critical nutrient for the enzymatic activity of proteins involved in myelination (Beard, Wiesinger, & Connor, Reference Beard, Wiesinger and Connor2003; Connor & Menzies, Reference Connor and Menzies1996), energy metabolism (Goodman, Warshaw, & Dallman, Reference Goodman, Warshaw and Dallman1970; Willis, Brooks, Henderson, & Dallman, Reference Willis, Brooks, Henderson and Dallman1987), and monoamine neurotransmission (Goodman et al., Reference Goodman, Warshaw and Dallman1970; Willis et al., Reference Willis, Brooks, Henderson and Dallman1987). The proteins include hydroxylases (e.g., tyrosine and tryptophan hydroxylase for dopamine and serotonin, respectively) and cytochromes that modulate electron transport in mitochondria.
The effects of fetal/neonatal ID follow the rules of timing, dose, and duration (Kretchmer et al., Reference Kretchmer, Beard and Carlson1996). Timing appears to be particularly important in determining the long-term behavioral phenotype following early ID. The prevalence of ID varies with age in the fetal and pediatric population. Three time periods are characterized by the highest prevalences: late fetal life, toddlerhood, and adolescence, particularly in menstruating females. ID in each of these epochs causes acute neurocognitive and motor effects (Geng et al., Reference Geng, Mai, Zhan, Xu, Zhao, Georgieff and Lozoff2015; Murray-Kolb & Beard, Reference Murray-Kolb and Beard2007; Siddappa et al., Reference Siddappa, Georgieff, Wewerka, Worwa, Nelson and Deregnier2004). However, only the two early time periods are associated with long-term effects in spite of iron repletion, suggesting that critical periods for brain structures exist in those early time periods. Consideration of which brain structures are rapidly developing when ID is present has informed our understanding of later childhood and adult behavioral phenotypes. For example, the late fetal period is characterized by rapid hippocampal development, onset of myelination, and sculpting of the dopamine neurotransmitter system. Fetal/neonatal ID compromises each of these, and behavioral phenotypes in humans are characterized by poorer declarative memory (Geng et al., Reference Geng, Mai, Zhan, Xu, Zhao, Georgieff and Lozoff2015; Siddappa et al., Reference Siddappa, Georgieff, Wewerka, Worwa, Nelson and Deregnier2004), slower speed of processing (Amin et al., Reference Amin, Orlando, Eddins, MacDonald, Monczynski and Wang2010; Roncagliolo, Garrido, Walter, Peirano, & Lozoff, Reference Roncagliolo, Garrido, Walter, Peirano and Lozoff1998), and alterations in mood and affect (Wachs et al., Reference Wachs, Pollitt, Cueto, Jacoby and Creed-Kanashiro2005). However, dopaminergic frontal lobe findings that are highly characteristic of ID in toddlerhood are not a prominent feature of fetal/neonatal ID most likely because of the relatively rudimentary nature of the prefrontal cortex.
Preclinical models confirm the regional brain vulnerability to fetal/neonatal ID. Two types of models are utilized to understand the neurobiology. The classic dietary maternal ID model in which the dam is started on an ID diet early in gestation with continuation until either Postnatal Day 10 (term human brain equivalent) or Postnatal Day 21 (weaning) has been the mainstay model (Felt et al., Reference Felt, Beard, Schallert, Shao, Aldridge, Connor and Lozoff2006; Rao et al., Reference Rao, Tkac, Townsend, Gruetter and Georgieff2003). More recently, genetically modified mice have been utilized to understand the role of iron in hippocampal neuronal development without the confounding factor of anemia (Carlson et al., Reference Carlson, Tkac, Magid, O'Connor, Andrews, Schallert and Petryk2009; Fretham et al., Reference Fretham, Carlson, Wobken, Tran, Petryk and Georgieff2012).
The bulk of the preclinical evidence in the literature is based on the dietary rat model. Gestational and gestational/lactational ID in rats alters brainwide fatty acid composition of myelin acutely during the period of deficiency (Connor & Menzies, Reference Connor and Menzies1996) and causes long-term suppression of myelin basic protein gene expression in adulthood after iron repletion (Clardy et al., Reference Clardy, Wang, Zhao, Liu, Chase, Beard and Connor2006). It alters dopamine and serotonin neurotransmission acutely in the neonatal period particularly in the nucleus accumbens, striatum, substantia nigra, and ventral midbrain. Mesocorticolimbic and nigrostriatal pathways are particularly vulnerable (Beard, Erikson, & Jones, Reference Beard, Erikson and Jones2003; Pinero, Jones, & Beard, Reference Pinero, Jones and Beard2001; Youdim & Green, Reference Youdim and Green1978). In adulthood, the cerebellum, frontal cortex, striatum, hippocampus, and midbrain show variations in markers of dopamine and serotonin metabolism, accompanied by alterations in behaviors that rely on these structures including negative geotaxis, forelimb placement, and novel object recognition (Unger et al., Reference Unger, Hurst, Georgieff, Schallert, Rao, Connor and Felt2012).
Fetal/neonatal dietary IDA in the rat or nonanemic neuronal-specific ID in the mouse alters adult hippocampal metabolism and neurotrophic factor gene expression (Carlson et al., Reference Carlson, Tkac, Magid, O'Connor, Andrews, Schallert and Petryk2009; Tran, Fretham, Carlson, & Georgieff, Reference Tran, Fretham, Carlson and Georgieff2009), dendrite morphology (Brunette et al., Reference Brunette, Tran, Wobken, Carlson and Georgieff2010; Carlson et al., Reference Carlson, Tkac, Magid, O'Connor, Andrews, Schallert and Petryk2009; Fretham et al., Reference Fretham, Carlson, Wobken, Tran, Petryk and Georgieff2012; Jorgenson, Wobken, & Georgieff, Reference Jorgenson, Wobken and Georgieff2003), electrophysiology (Jorgenson et al., Reference Jorgenson, Sun, O'Connor and Georgieff2005; Pisansky et al., Reference Pisansky, Wickham, Su, Fretham, Yuan, Sun and Georgieff2013), and learning and memory behavior (Carlson et al., Reference Carlson, Tkac, Magid, O'Connor, Andrews, Schallert and Petryk2009; Felt & Lozoff, Reference Felt and Lozoff1996; Fretham et al., Reference Fretham, Carlson, Wobken, Tran, Petryk and Georgieff2012; Schmidt, Waldow, Grove, Salinas, & Georgieff, Reference Schmidt, Waldow, Grove, Salinas and Georgieff2007). Persistence of these effects into adulthood in the dietary rat model occurred in spite of initiation of iron treatment during the lactation phase (Unger et al., Reference Unger, Hurst, Georgieff, Schallert, Rao, Connor and Felt2012). In the specifically neuronal ID model, adult structural and behavioral changes were prevented when iron was repleted in the neurons during lactation (Fretham et al., Reference Fretham, Carlson, Wobken, Tran, Petryk and Georgieff2012). The likely cause of the discrepancy between the dietary rat and genomic mouse models is that the rat is anemic and total body iron deficient and thus the repletion period is much longer compared to the genomic mouse model. The long-term findings in both models suggest a critical period for iron during development, most likely during the rapid brain growth phase immediately postnatally.
The adult behavioral phenotypes seen in both rodent species models are instructive with respect to developmental psychopathology. An important finding from the nonanemic hippocampal neuronal ID mouse models was that extrahippocampal abnormalities were also noted in the striatum in spite of normal iron content (Carlson et al., Reference Carlson, Fretham, Unger, O'Connor, Petryk, Schallert and Georgieff2010) in addition to the spatial learning and memory deficits that would be expected from a hippocampal iron deficit (Carlson et al., Reference Carlson, Tkac, Magid, O'Connor, Andrews, Schallert and Petryk2009; Fretham et al., Reference Fretham, Carlson, Wobken, Tran, Petryk and Georgieff2012). Specifically, striatal metabolism is compromised by hippocampal ID along with procedural memory behaviors that depend on an intact striatum (Carlson et al., Reference Carlson, Fretham, Unger, O'Connor, Petryk, Schallert and Georgieff2010). The findings emphasize the interconnectedness of these brain areas as they work together in integrated, complex neural circuits. The striatal findings may well be driven by compromised hippocampal-derived glutamatergic neurons (Jorgenson et al., Reference Jorgenson, Sun, O'Connor and Georgieff2005) failing to normally potentiate dopaminergic neurons in the VTA loop. Similar circuit effects were demonstrated by Pisansky et al. (Reference Pisansky, Wickham, Su, Fretham, Yuan, Sun and Georgieff2013) in the nonanemic hippocampal neuron specific and the dietary IDA models (Pisansky et al., Reference Pisansky, Wickham, Su, Fretham, Yuan, Sun and Georgieff2013). Paired-pulse inhibition (PPI), a measure of sensory gating often utilized as a model of schizophrenia, is dependent on normal hippocampal function in the complex circuitry that underlies this behavior. PPI is highly abnormal in the adult mouse and rat following fetal/neonatal ID, suggesting that hippocampal damage sustained in the neonatal period continues to have ramifications at the circuit level. The finding also suggests a plausible biology for the epidemiologic finding that fetal ID increases the risk of schizophrenia in adulthood in humans (Insel et al., Reference Insel, Schaefer, McKeague, Susser and Brown2008).
Schmidt et al. (Reference Schmidt, Waldow, Grove, Salinas and Georgieff2007, Reference Schmidt, Alvarez, Grove, Rao and Georgieff2012) also found evidence of circuit-level dysfunction in adulthood following fetal/neonatal dietary ID in the rat. Formerly ID adult rats showed expected impairments in spatial memory (Schmidt et al., Reference Schmidt, Waldow, Grove, Salinas and Georgieff2007), but superior performance on set-shifting paradigms (Schmidt et al., Reference Schmidt, Alvarez, Grove, Rao and Georgieff2012). The spatial memory abnormalities are consistent with the anatomic and electrophysiologic studies in the model that showed permanent hippocampal damage (Brunette et al., Reference Brunette, Tran, Wobken, Carlson and Georgieff2010; Jorgenson et al., Reference Jorgenson, Sun, O'Connor and Georgieff2005). However, the increased cognitive flexibility seen in the set-shifting paradigm implies intact or enhanced frontal lobe function. The lower amount of hippocampal inhibition allowed the rats to utilize frontal strategies to successfully complete the set-shifting paradigm. This finding illustrates an important circuitry principle because the hippocampus and frontal cortex coordinately regulate VTA loop activity. Disruption of the balance of these inputs is thought to underlie the disordered decision making observed in schizophrenia (Belujon & Grace, Reference Belujon and Grace2008; Lisman et al., Reference Lisman, Coyle, Green, Javitt, Benes, Heckers and Grace2008). Selective lesioning of either hippocampal or frontal inputs results in a schizophrenia phenotype in the rodent (Esmaeili & Grace, Reference Esmaeili and Grace2013).
Nonhuman primates have been used in a small number of studies to demonstrate the importance of timing of ID in a more humanlike model. Prenatal ID results in an impulsive behavioral phenotype in the juvenile animal, while postnatal ID results a hesitant, wary phenotype (Golub, Reference Golub2010). The findings are consistent with those in humans. Felt et al. (Reference Felt, Beard, Schallert, Shao, Aldridge, Connor and Lozoff2006) have described a hesitant/wary behavioral phenotype in postnatally ID animals and have postulated that the effects are due to compromise of dopaminergic projections to the frontal lobe from the striatum. In contrast, the ADHD phenotype seen with prenatal ID may be due to a secondary compromise of hippocampal-derived projections to the prefrontal cortex
As noted previously, two potential principles can explicate these long-term negative effects. First, in terms of a structure–function relationship, poorly formed structure can result in poor neural plasticity and gene regulation. Classical neurotransmitters such as dopamine, whose synthesis is dependent on iron-dependent tyrosine hydroxylase, can compromise neuron number in the developing cortex (Kolk et al., Reference Kolk, Gunput, Tran, van den Heuvel, Prasad, Hellemons and Pasterkamp2009; Popolo, McCarthy, & Bhide, Reference Popolo, McCarthy and Bhide2004). Second, emerging evidence suggests fetal/neonatal ID also alters the function of iron-dependent epigenetic modifiers, leading to stable changes in chromatin remodeling, providing an alternative mechanism underlying the long-term gene dysregulation in neurons (Tran et al., Reference Tran, Kennedy, Lien, Simmons and Georgieff2015).
In preclinical models, early life IDA permanently alters the regulation of genes that are important for adult function, including neurological networks underlying synaptic plasticity, major depression, anxiety, autism, and schizophrenia (Brunette et al., Reference Brunette, Tran, Wobken, Carlson and Georgieff2010; Carlson, Stead, Neal, Petryk, & Georgieff, Reference Carlson, Stead, Neal, Petryk and Georgieff2007; Patton, Coe, Lubach, & Connor, Reference Patton, Coe, Lubach and Connor2012; Tran, Dakoji, Reise, Storey, & Georgieff, Reference Tran, Dakoji, Reise, Storey and Georgieff2013; Tran et al., Reference Tran, Kennedy, Pisansky, Won, Gewirtz, Simmons and Georgieff2016). Epigenetic modifications of these gene networks by iron are highly plausible mechanisms to explain these long-term findings because two of the known epigenetic mechanisms (i.e., methyl-cytosine hydroxylation and histone methylation) are iron dependent and are mediated by two families of iron-containing proteins: the ten-eleven-translocation methyl-cytosine hydroxylases (TETs) and the methyl-lysine demethylases (KDMs and JARIDs). Both protein families require iron for their catalytic activity (Kouzarides, Reference Kouzarides2007; Pedersen & Helin, Reference Pedersen and Helin2010; Ponnaluri, Maciejewski, & Mukherji, Reference Ponnaluri, Maciejewski and Mukherji2013). KDMs interact with histone deacetylases (Barrett et al., Reference Barrett, Santangelo, Tan, Catchpole, Roberts, Spencer-Dene and Taylor-Papadimitriou2007; Chang, Chen, Zhao, & Bruick, Reference Chang, Chen, Zhao and Bruick2007; Chicas et al., Reference Chicas, Kapoor, Wang, Aksoy, Evertts, Zhang and Lowe2012; Ramadoss, Chen, & Wang, Reference Ramadoss, Chen and Wang2012; Suh, Aoyama, Matsumori, Liu, & Swanson, Reference Suh, Aoyama, Matsumori, Liu and Swanson2005), further supporting a mechanistic link between neuronal iron status and epigenetic modifications. The findings of fetal/neonatal ID effects on TET activity remain unknown. In contrast, the persistently dysregulated KDMs accompanied by substantial changes in histone methylation at the Bdnf locus (Blegen et al., Reference Blegen, Kennedy, Thibert, Gewirtz, Tran and Georgieff2013; Tran et al., Reference Tran, Kennedy, Lien, Simmons and Georgieff2015), suggesting KDM dysfunction in the adult hippocampus following early life ID, provides a strong rationale for KDM's role in reprogramming gene regulation. In addition to the intracellular iron content, KDMs may be further regulated by iron-dependent proline hydroxylases (PHDs), thereby potentiating the effect of early life ID. Iron availability is essential for PHD to hydroxylate proline residues of HIF1a (Kaelin & Ratcliffe, Reference Kaelin and Ratcliffe2008; Nandal et al., Reference Nandal, Ruiz, Subramanian, Ghimire-Rijal, Sinnamon, Stemmler and Philpott2011), which in turn directly regulates the expression of KDMs (Beyer, Kristensen, Jensen, Johansen, & Staller, Reference Beyer, Kristensen, Jensen, Johansen and Staller2008; Lee, Choi, Oh, Park, & Park, Reference Lee, Choi, Oh, Park and Park2014; Mudie et al., Reference Mudie, Bandarra, Batie, Biddlestone, Moniz, Ortmann and Rocha2014; Niu et al., Reference Niu, Zhang, Liao, Zhou, Lindner, Zhou and Yang2012; Pollard et al., Reference Pollard, Loenarz, Mole, McDonough, Gleadle, Schofield and Ratcliffe2008). PHD1 expression is upregulated acutely in ID hippocampus (Carlson et al., Reference Carlson, Stead, Neal, Petryk and Georgieff2007) and is normalized following iron treatment in the formerly ID hippocampus (Tran et al., Reference Tran, Kennedy, Pisansky, Won, Gewirtz, Simmons and Georgieff2016). The finding that KDMs remain dysregulated while PHD effect is resolved in adulthood points to a role of PHD in reprogramming of KDMs during the neonatal period. Elucidating the iron-dependent epigenetic mechanisms and their roles in reprogramming neural genes remains an emerging field and an active area of research.
Epidemiological and clinical studies of IUGR
IUGR occurs in 10% of pregnancies. Worldwide, the most common etiology of IUGR is maternal undernutrition, while in the United States the most common cause is nutrient restriction due to placental insufficiency from maternal hypertension. Maternal infections particularly early in the pregnancy also cause IUGR. Each would be expected to have differential effects on the developing brain, and animal models can provide such insight. All etiologies result in restriction of nutrient flow from mother to fetus and result in a fetus that is smaller than its genetic potential. Both maternal malnutrition and IUGR have been epidemiologically associated with increased risks of developmental psychopathologies, including schizophrenia, autism, depression, and neurocognitive delay.
The relationship of IUGR to schizophrenia has been documented in several studies (Susser et al., Reference Susser, Neugebauer, Hoek, Brown, Lin, Labovitz and Gorman1996; Susser & Lin, Reference Susser and Lin1992). A meta-analysis revealed that birth weight <2000 g and >2500g is significantly associated with development of schizophrenia (Cannon et al., Reference Cannon, Jones and Murray2002). Not only is a birth weight of less than 2500g at term significantly associated with increased risk of schizophrenia, but a significant linear trend of increasing odds ratios with decreasing birth weight has been observed for infants with birth weights greater than 2500g (Abel et al., Reference Abel, Wicks, Susser, Dalman, Pedersen, Mortensen and Webb2010). Of note, the same risk pattern has been shown for other psychiatric categories, including any psychiatric diagnosis; alcohol and drug disorders; affective disorders; and neurotic, stress-related, and somatoform disorders (Abel et al., Reference Abel, Wicks, Susser, Dalman, Pedersen, Mortensen and Webb2010).
The risk of autism is associated with daily smoking during pregnancy (a risk factor for IUGR), and with being low weight for gestational age (Hultman, Sparen, & Cnattingius, Reference Hultman, Sparen and Cnattingius2002). Depression in adulthood has also been associated with having had IUGR as a fetus. Young adults, who were born very low birth weight and small for gestational age reported significantly higher use of antidepressants, higher scores on the Beck Depression Inventory, and more likely to report a depression diagnosis than control subjects and very low birth weight infants born appropriate for gestational age (AGA; Raikkonen et al., Reference Raikkonen, Pesonen, Heinonen, Kajantie, Hovi, Jarvenpaa and Andersson2008). A prospective study of IUGR infants born preterm compared with age-match preterm AGA and term AGA controls found decreased verbal and full-scale IQ scores; IUGR and preterm AGA infants had lower performance IQ than term control subjects (Morsing, Asard, Ley, Stjernqvist, & Marsal, Reference Morsing, Asard, Ley, Stjernqvist and Marsal2011). Small body size at birth significantly correlates with increased symptoms of ADHD in children ages 5 to 6 years old (Lahti et al., Reference Lahti, Raikkonen, Kajantie, Heinonen, Pesonen, Jarvenpaa and Strandberg2006). Being small for gestational age and born preterm is significantly correlated with risk of psychiatric hospitalization for drug and alcohol dependency in adulthood (Nosarti et al., Reference Nosarti, Reichenberg, Murray, Cnattingius, Lambe, Yin and Hultman2012). Placental pathology consistent with underperfusion, a hallmark of IUGR, is associated with poorer cognitive, language, and motor performance at 2 years of age in near-term small for gestational age infants (Parra-Saavedra et al., Reference Parra-Saavedra, Crovetto, Triunfo, Savchev, Peguero, Nadal and Figueras2014).
Neuroimaging studies confirm the global and regional effects of IUGR on neuroanatomy seen in preclinical models. Globally, IUGR infants have reductions in absolute cortical gray matter volume and overall brain tissue volume, and lack of postnatal catch-up brain growth. The anatomic findings correlate with reductions in attention and interaction capacity at term, and less mature neurobehavioral development on the Assessment of Preterm Infant Behavior (Tolsa et al., Reference Tolsa, Zimine, Warfield, Freschi, Sancho Rossignol, Lazeyras and Huppi2004).
Several studies have found changes in regional brain size of IUGR infants. IUGR infants have reductions in frontal lobe and cerebellar volumes (Caetano et al., Reference Caetano, Zamarian, Araujo Junior, Cavalcante, Simioni, Silva and Nardozza2015) as well as reduction in hippocampal volume on MRI (Lodygensky et al., Reference Lodygensky, Seghier, Warfield, Tolsa, Sizonenko, Lazeyras and Huppi2008). Fetuses with late-onset IUGR had deeper measurements of left and right insula and cingulate fissure as well as smaller intracranial, total brain, left opercula, and right opercula volumes (Egana-Ugrinovic, Sanz-Cortes, Figueras, Bargallo, & Gratacos, Reference Egana-Ugrinovic, Sanz-Cortes, Figueras, Bargallo and Gratacos2013). In another study, IUGR fetuses had reductions in brain volume, but no demonstrable delay in cortical maturation based on grading of sulci and gyri; instead, they showed evidence of accelerated cortical development on these measures (Businelli, de Wit, Visser, & Pistorius, Reference Businelli, de Wit, Visser and Pistorius2014). IUGR infants had texture changes in several brain regions including the frontal cortex, cingulum, basal ganglia, midbrain, and cerebellum, indicative of microstructural differences and possibly brain reorganization compared to AGA infants (Sanz-Cortes et al., Reference Sanz-Cortes, Figueras, Bonet-Carne, Padilla, Tenorio, Bargallo and Gratacos2013). IUGR altered markers of maturation in frontal white matter, thalami, centrum semiovale, and pons associated with cognitive disorders in 36% of children at follow-up (Arthurs et al., Reference Arthurs, Rega, Guimiot, Belarbi, Rosenblatt, Biran and Alison2017). A tractography, a 3-D modeling of neural track using diffusion tensor imaging data, study showed that IUGR is associated with altered connectivity in motor and cortico–striatal–thalamic networks (Eixarch, Munoz-Moreno, Bargallo, Batalle, & Gratacos, Reference Eixarch, Munoz-Moreno, Bargallo, Batalle and Gratacos2016). A stereological study of postmortem brain tissue of IUGR infants found severe reduction of total cell numbers in the cortex, but not in other developmental zones (Samuelsen et al., Reference Samuelsen, Pakkenberg, Bogdanovic, Gundersen, Larsen, Graem and Laursen2007). A brain network topology study showed evidence of brain reorganization in IUGR infants at 1 year of age (Batalle et al., Reference Batalle, Eixarch, Figueras, Munoz-Moreno, Bargallo, Illa and Gratacos2012). IUGR infants had decreases in measures of global and local connectivity, which were associated with abnormal performance in later cognitive, language, and motor outcomes at 2 years of age (Batalle et al., Reference Batalle, Eixarch, Figueras, Munoz-Moreno, Bargallo, Illa and Gratacos2012). Structural MRI showed that 12-month-old IUGR infants had gray matter reductions in temporal, parietal, frontal, and insular regions; increased white matter in temporal regions compared to the AGA group and in frontal, parietal, occipital, and insular regions compared to term group; and decreased white matter in the cerebellum compared to term infants (Padilla et al., Reference Padilla, Falcon, Sanz-Cortes, Figueras, Bargallo, Crispi and Gratacos2011). Functional consequences at the electrophysiologic and behavioral levels occur as a function of degree of IUGR. IUGR results in markers of delayed maturation of EEG, and associated delay in neuromotor development (Yerushalmy-Feler et al., Reference Yerushalmy-Feler, Marom, Peylan, Korn, Haham, Mandel and Bassan2014). Poorer intellectual performance at 7 years of age is associated when poor weight gain occurs postnatally in infants with IUGR (Pylipow et al., Reference Pylipow, Spector, Puumala, Boys, Cohen and Georgieff2009), indicating that sensitive periods for brain growth and connectivity to nutrient availability extend beyond the fetal period.
Preclinical models and molecular mechanisms of IUGR
IUGR is not a single nutrient disorder; rather, it represents the anthropometric end result of global fetal malnutrition. Placental transfer of nutrients is the final common pathway from the mother to the developing fetus, and disruption of that process by any number of conditions results in the “unifinality” of retarded or restricted intrauterine growth. The failure to grow reflects primarily macronutrient (i.e., protein and calorie) malnutrition although micronutrient deficits (e.g., iron ID) are also prevalent and can restrict somatic and brain growth (Chockalingam, Murphy, Ophoven, Weisdorf, & Georgieff, Reference Chockalingam, Murphy, Ophoven, Weisdorf and Georgieff1987; Georgieff, Mills, Gordon, & Wobken, Reference Georgieff, Mills, Gordon and Wobken1995). The traditional assessment of growth restriction through anthropometric analysis of weight, length, and head circumference for estimated gestational age is a blunt tool that does not have the precision to detect differential effects of the various etiologies of growth restriction on the developing brain.
Multiple preclinical models of IUGR have been developed because of the intense interest in Barker's observation of “fetal programming” of adult cardiovascular disease (Barker, Reference Barker1997; Gluckman & Hanson, Reference Gluckman and Hanson2004). Typically, rodent models have predominated because their short life spans allow for assessment of adult outcomes following fetal nutrient restriction events. The two most commonly used models are maternal dietary restriction and uterine artery (UA) ligation. Maternal dietary restriction is usually accomplished through reducing total food consumption during the entire pregnancy, thus shortchanging the mother of macro- and micronutrients. This approach models severe malnutrition that often presents in the “majority world.” Individual nutrients such as protein or energy (calories) are restricted in some experiments while we maintain the rest of the diet in order to isolate the specific role of the nutrient in question. UA ligation in late gestation accomplishes a similar fetal nutrient restriction as the dietary models but does so without compromising maternal health status. It models the IUGR that occurs as a consequence of gestational hypertension in an otherwise nutritionally sufficient mother, seen primarily in “Westernized” countries.
Preclinical models other than rodents (e.g., sheep) have been used to assess the effects of IUGR predominantly on fetal and neonatal metabolism in order to provide information about insulin resistance and cardiovascular risk (Hay, Brown, Rozance, Wesolowski, & Limesand, Reference Hay, Brown, Rozance, Wesolowski and Limesand2016). However, sheep are limited in the information they can provide about brain development and risk of later psychopathology. Rodents are the preferred model for studying IUGR on brain development because of their robust behavioral repertoire and the analogies that can be drawn between their abnormalities and those of humans (Table 2).
Table 2. Integrated approaches between preclinical models and humans that provide biological plausibility in assessment of nonfatal developmental disorders
Note: ERP, event-related potential. MRS, magnetic resonance spectroscopy. CSF, cerebrospinal fluid.
All nutrients are needed for normal brain development. As IUGR preclinical models typically restrict global nutrient delivery from mother to fetus, global effects are observed in the brain. Differential regional effects, where one brain area may be more affected than another, are largely related to relative metabolic rates (and thus nutrient demands) of the affected regions with those regions with higher metabolic demands at the time of the nutrient restriction being most affected. Regional metabolic demands are driven by growth rates, which in turn are driven by the age of the fetus. In humans, malnutrition early in pregnancy (or throughout pregnancy) results in global loss of brain volume and microcephaly (Winick, Reference Winick1971). The microcephaly in humans is due to fewer cells and lower amounts of DNA and RNA (Rosso, Hormazabal, & Winick, Reference Rosso, Hormazabal and Winick1970; Winick, Reference Winick1971; Winick, Rosso, & Waterlow, Reference Winick, Rosso and Waterlow1970). First trimester viral infectious agents such as cytomegalovirus also restrict neurogenesis and result in microcephaly (Kagan & Hamprecht, Reference Kagan and Hamprecht2017; Swanson & Schleiss, Reference Swanson and Schleiss2013). Migration defects are more prominent with earlier nutritional abnormalities.
In contrast, nutrient deficiencies that occur later in gestation have less global effects on neurogenesis, but more intense effects on neural differentiation in regions that are developing just prior to birth (e.g., hippocampus). Selective disruption of regional development can contribute to unbalanced neural circuit function later in life.
An extensive meta-analysis of the literature on adult neurodevelopmental outcomes following UA ligation in rats, mice, and guinea pigs was recently reviewed by Basilious, Yager, and Fehlings (Reference Basilious, Yager and Fehlings2015). Global effects were noted in all of the models specifically related to the degree of somatic growth restriction. Ventricular enlargement, indicative of reduced brain volume, was common across species (Mallard, Rehn, Rees, Tolcos, & Copolov, Reference Mallard, Rehn, Rees, Tolcos and Copolov1999; Rehn et al., Reference Rehn, van den Buuse, Copolov, Briscoe, Lambert and Rees2004; Ruff et al., Reference Ruff, Faulkner, Rumajogee, Beldick, Foltz, Corrigan and Fehlings2017). The loss of brain volume is due to compromise of both white and gray matter. Oligodendrocyte precursors are reduced throughout the brain, resulting in a delay in onset of myelination (Olivier, Baud, Evrard, Gressens, & Verney, Reference Olivier, Baud, Evrard, Gressens and Verney2005; Reid et al., Reference Reid, Murray, Marsh, Golden, Simmons and Grinspan2012) and an overall loss of myelination in adulthood (Ruff et al., Reference Ruff, Faulkner, Rumajogee, Beldick, Foltz, Corrigan and Fehlings2017). Major white matter tracts are reduced in volume and density (Delcour, Olivier, et al., Reference Delcour, Olivier, Chambon, Pansiot, Russier, Liberge and Coq2012; Delcour, Russier, et al., Reference Delcour, Russier, Amin, Baud, Paban, Barbe and Coq2012; Ruff et al., Reference Ruff, Faulkner, Rumajogee, Beldick, Foltz, Corrigan and Fehlings2017). The loss of white matter can be widespread, including within the corpus callosum, brainstem, hippocampus, and somatosensory cortex (Delcour, Olivier, et al., Reference Delcour, Olivier, Chambon, Pansiot, Russier, Liberge and Coq2012). The profound effects on white matter are not surprising because IUGR compromises maternal–fetal oxygen transport and oligodendrocytes are highly sensitive to hypoxia.
Generalized gray matter volume loss in IUGR models occurs predominantly from global and regional loss of neurons through neuronal cell death and/or apoptosis (Delcour, Olivier, et al., Reference Delcour, Olivier, Chambon, Pansiot, Russier, Liberge and Coq2012; Mallard, Loeliger, Copolov, & Rees, Reference Mallard, Loeliger, Copolov and Rees2000). Beyond reduced neuronal numbers, dendrite branching patterns are less complex and contribute to the volume loss particularly in the hippocampus (Dieni & Rees, Reference Dieni and Rees2003). Neuronal function is clearly related to dendrite branching complexity; thus, the loss of complexity due to the substrate restriction of IUGR would be expected to compromise behaviors that are dependent on the affected structures. Neuronal loss and loss of morphological integrity is found in all hippocampal areas (Mallard et al., Reference Mallard, Loeliger, Copolov and Rees2000; Ruff et al., Reference Ruff, Faulkner, Rumajogee, Beldick, Foltz, Corrigan and Fehlings2017), the caudate nucleus (Rehn et al., Reference Rehn, van den Buuse, Copolov, Briscoe, Lambert and Rees2004; Ruff et al., Reference Ruff, Faulkner, Rumajogee, Beldick, Foltz, Corrigan and Fehlings2017), the cerebral cortex (Mallard et al., Reference Mallard, Rehn, Rees, Tolcos and Copolov1999), the medial and entorhinal cortex (Basilious et al., Reference Basilious, Yager and Fehlings2015), and the cerebellum (Mallard et al., Reference Mallard, Loeliger, Copolov and Rees2000).
Beyond the structural changes, IUGR due to UA ligation also compromises signaling pathways that are critical for establishment and maintenance of synaptic plasticity. BDNF expression is reduced throughout the brain (Nishigori, Mazzuca, Nygard, Han, & Richardson, Reference Nishigori, Mazzuca, Nygard, Han and Richardson2008) and specifically in the hippocampus (Dieni & Rees, Reference Dieni and Rees2005). The hippocampus is highly dependent on glutamatergic signaling, which is disrupted significantly by IUGR. NMDA receptor subunit expression is reduced overall, and a delay in the maturational switch of subunit NR2a to NR2b occurs (Catteau et al., Reference Catteau, Gernet, Marret, Legros, Gressens, Leroux and Laudenbach2011; Schober et al., Reference Schober, McKnight, Yu, Callaway, Ke and Lane2009). Neuroprotective pathways in the cortex and hippocampus are compromised as evidenced by an increase in the pro-apoptotic p53 protein and a reduction in the anti-apoptotic Bcl-2 protein (Ke et al., Reference Ke, McKnight, Wang, Yu, Wang, Callaway and Lane2005; Lane et al., Reference Lane, Ramirez, Tsirka, Kloesz, McLaughlin, Gruetzmacher and Devaskar2001).
Overall, preclinical models of late fetal IUGR due to placental insufficiency indicate widespread acute abnormalities, many of which carry forward into adulthood. The changes are found at the anatomic and signaling levels and are particularly prominent in regions that are rapidly developing and demonstrate critical periods of development in the late fetal/early neonatal stages. UA ligation results in adult behavioral abnormalities that rely on the regions that were affected in the neonatal period. Specifically, cerebellar, hippocampal, and cortical injury due to IUGR results in abnormalities of gait, memory, object recognition, and spatial processing in adulthood (Basilious et al., Reference Basilious, Yager and Fehlings2015).
In considering the epidemiologic data in humans linking IUGR to developmental psychopathologies that emerge later in life, it is important to note that all of the fetally affected regions are destined to be interconnected postnatally to establish functional circuits underlying far more complex behaviors. Thus, a multiplicative or ripple effect throughout the nervous system likely occurs through complex circuits (e.g., the VTA loop or mesolimbic frontal connections), thus providing the anatomic framework for abnormalities in attentional regulation, ordered thinking, cognitive flexibility, sensory gating, and set-shifting behaviors that have been noted in adult survivors of IUGR. For example, rats with UA ligation-induced IUGR have damage to hippocampal area CA1 and prefrontal cortex and demonstrate hyperactivity (Delcour, Olivier, et al., Reference Delcour, Olivier, Chambon, Pansiot, Russier, Liberge and Coq2012; Delcour, Russier, et al., Reference Delcour, Russier, Amin, Baud, Paban, Barbe and Coq2012) similar to the ADHD phenotype described in humans. Prepulse inhibition abnormalities have been noted in the guinea pig model of IUGR (Rehn et al., Reference Rehn, van den Buuse, Copolov, Briscoe, Lambert and Rees2004). Impaired PPI has been utilized as a marker of the sensory gating abnormalities in schizophrenia (Rehn et al., Reference Rehn, van den Buuse, Copolov, Briscoe, Lambert and Rees2004), the incidence of which is increased in adults following IUGR. The behavioral studies in the preclinical models provide biological plausibility for primary (e.g., learning and memory deficits) as well as downstream complex behavioral abnormalities (i.e., sensory gating) noted in humans following IUGR.
Similar to the effects of ID, the role of timing in driving the acute and long-term abnormalities should be appreciated with IUGR. Late onset IUGR, as seen with the late gestation UA ligation model, induces severe, but relatively short-lived reductions in critical nutritional/metabolic substrates. This “one-hit” model produces a large “area under the curve” effect more from the large dose of the insult, as opposed to its duration (Kretchmer et al., Reference Kretchmer, Beard and Carlson1996). An encephalopathic insult relegated to a brief epoch significantly damages only those regions with the greatest nutrient demand. Regions that have a more protracted critical period of development (and that show large amounts of plasticity) theoretically have an opportunity to recover, while those with relatively brief critical periods (and lesser plasticity) likely end up with permanent effects. Studying the effect of early versus late onset fetal UA ligation is technically challenging in the rodent due to excessive fetal demise in the early treatment group. However, the question has been studied in fetal sheep and supports the principle that timing matters. Second-trimester UA ligation in sheep results in more widespread white matter injury and neuroinflammation in the cortex when compared to third-trimester UA ligation (Alves de Alencar Rocha et al., Reference Alves de Alencar Rocha, Allison, Yawno, Polglase, Sutherland, Malhotra and Miller2017). The greater induction of inflammation during a period of extensive neuronal migration is particularly concerning, especially in light of recent theories that ASD may have a neuroinflammatory etiology.
In contrast to late-gestation UA ligation experiments, IUGR due to chronic maternal malnutrition throughout pregnancy results in an “area under the curve” characterized by a low dose insult over a long duration. While the magnitude of the Dose × Duration area under the curve may be similar to UA ligation, the shape is decidedly different, with many more periods of critical regional development at risk. The effects of IUGR due to maternal malnutrition are more global than UA ligation.
Macronutrient IUGR models restrict protein or protein + energy intake of the mother beginning early in the first trimester. The nutrient restriction is present throughout pregnancy and is frequently severe with the dams receiving as little as 50% of their normal intake. The effect of protein restriction at various stages of pregnancy has been described in rodent models since 1975 (Koshy, Sara, King, & Lazarus, Reference Koshy, Sara, King and Lazarus1975).
The global findings in these preclinical models corroborate the findings from autopsy studies in human infants including a reduction in brain size due primarily to loss of neuronal number (Rosso et al., Reference Rosso, Hormazabal and Winick1970; Winick, Reference Winick1971). The models suggest that maternal malnutrition affects the rate of brain development by causing premature cessation of neurogenesis and an earlier switch from proliferation to cell differentiation (Koshy et al., Reference Koshy, Sara, King and Lazarus1975). IUGR induced by a low-protein diet given to the rat dam starting on Day 1 of pregnancy reduces overall myelination and connectivity in the offspring. Significant deregulation of genes controlling neuroinflammation occurs, allowing for a more pro-inflammatory state in the developing brain (Rideau Batista Novais et al., Reference Rideau Batista Novais, Pham, van de Looij, Bernal, Mairesse, Zana-Taieb and Baud2016). The authors suggest that neuroinflammation due to IUGR may provide a biological explanation for poor cognitive outcome in IUGR humans (Rideau Batista Novais et al., Reference Rideau Batista Novais, Pham, van de Looij, Bernal, Mairesse, Zana-Taieb and Baud2016).
As with UA ligation, regional brain effects are also seen with protein and protein-energy restriction. The hypothalamus is reprogrammed with respect to satiety and hunger cues, which may relate to appetite regulation issues and metabolic syndrome seen in adulthood following IUGR (Pedroso et al., Reference Pedroso, Souza, Dornellas, Oyama, Nascimento, Santos and Ribeiro2017). IUGR also lowers cerebral weight, resulting in fewer neurons through increased apoptosis accompanied by reduction in PKA-CREB signaling pathway activity (Liu, Liu, & Chen, Reference Liu, Liu and Chen2011; Liu, Liu, Wang, Chen, & Yang, Reference Liu, Liu, Wang, Chen and Yang2013). Synaptic counts are reduced, and glial cell proliferation is suppressed in the cortex (Liu et al., Reference Liu, Liu and Chen2011). IUGR increases oxidative stress and results in lipid peroxidation in the cerebellum and the cerebral cortex (Tatli et al., Reference Tatli, Guzel, Kizil, Kavak, Yavuz and Kizil2007).
Behaviorally, the long-term effects are particularly magnified if malnutrition continues postnatally after IUGR. Rat pups born to dams on a low-protein diet and maintained on that diet to adulthood have poor spatial learning and memory. Male offspring have more hyperactivity than females (Naik, Patro, & Patro, Reference Naik, Patro and Patro2015). IUGR followed by postnatal growth restriction increases anxiety in males and females in young adulthood, but without social or cognitive impairment. Nevertheless, both males and females exhibit increased biochemical evidence of Alzheimer disease at 15–17 months with more astrogliosis and a tenfold increase in amyloid-beta42 protein expression (Tomi, Zhao, Thamotharan, Shin, & Devaskar, Reference Tomi, Zhao, Thamotharan, Shin and Devaskar2013).
Studies of both IUGR human cohorts and animal models have provided evidence for epigenetic programming of the epigenome (Heijmans et al., Reference Heijmans, Tobi, Lumey and Slagboom2009; Joss-Moore, Albertine, & Lane, Reference Joss-Moore, Albertine and Lane2011; Simmons, Reference Simmons2007; Tobi et al., Reference Tobi, Lumey, Talens, Kremer, Putter, Stein and Heijmans2009). DNA methylation was examined at the regulatory regions of 15 genes associated with cardiovascular and metabolic disease in adults who were exposed to famine (members of the Dutch famine cohort) during gestation. Gestational famine significantly altered the DNA methylation at 6 of these 15 gene regulatory loci, indicating that gene expression might be altered in these individuals, even in adulthood, long after the period of gestational famine (Heijmans et al., Reference Heijmans, Tobi, Lumey and Slagboom2009; Tobi et al., Reference Tobi, Lumey, Talens, Kremer, Putter, Stein and Heijmans2009). Both modifications of DNA and histones have been demonstrated in IUGR animal models, particularly in genes that have important roles in brain development and brain health across the life span (Joss-Moore et al., Reference Joss-Moore, Albertine and Lane2011; Simmons, Reference Simmons2007).
Concluding Remarks
Clinically, psychopathologies are thought of as discrete entities (as found in the DSM-5 or ICD-10) with little overlap, and their diagnosis points toward specific treatment protocols (Caspi et al., Reference Caspi, Houts, Belsky, Goldman-Mellor, Harrington, Israel and Moffitt2014). Practically, there can be difficulty even with experienced clinicians in distinguishing between presentations of different psychiatric illnesses, as diagnosis often entirely relies on presenting signs and symptoms, without regard to the underlying neurobiology (Cuthbert & Insel, Reference Cuthbert and Insel2013). This is especially true in children, where comorbidity of different psychopathologies is more common than in adults (Angold, Costello, & Erkanli, Reference Angold, Costello and Erkanli1999). This is especially notable in that genes identified as risk factors (via genome-wide association studies in this reference) in multiple psychopathologies have remarkable overlap (Cross-Disorder Group of the Psychiatric Genomics Consortium, 2013), and meta analyses of imaging studies have found neural substrates common for both individual psychiatric entities (an affective subdivision of the corticostriatal–pallidal–thalamic circuit in depression; Hamilton et al., Reference Hamilton, Etkin, Furman, Lemus, Johnson and Gotlib2012) and common to multiple psychiatric diagnoses (i.e., transdiagnostically across different psychopathologies; gray matter loss in the dorsal anterior cingulate, right insula, and left insula; Goodkind et al., Reference Goodkind, Eickhoff, Oathes, Jiang, Chang, Jones-Hagata and Etkin2015). These imaging studies further point toward neural substrates supporting specific behaviors (increased salience of negative information in major depressive disorder and a transdiagnostic association with poorer executive functioning). A fundamental question with respect to psychopathology resulting from events of commission or omission in the intrauterine environment is whether these are purely environmental influences or if they represent an interaction between gene and environment, which could be mistaken for either genetic traits or environmental insults. For example, evidence from twin studies demonstrates a large correlation between internalizing and externalizing factors, with a strong genetic influence (Cosgrove et al., Reference Cosgrove, Rhee, Gelhorn, Boeldt, Corley, Ehringer and Hewitt2011; Lahey, van Hulle, Singh, Waldman, & Rathouz, Reference Lahey, van Hulle, Singh, Waldman and Rathouz2011). Internalizing behaviors are considered “overcontrolled” behaviors characterized by anxiety and depression, whereas externalizing behaviors are considered “undercontrolled” and characterized by aggression and hyperactivity (Achenbach & Edelbrock, Reference Achenbach and Edelbrock1984). In this same study, psychiatric disorder-specific causes were mostly attributed to nonshared environmental influences. These results are consistent with a “generalist gene, specialist environments model” of psychopathology (Eley, Reference Eley1997). Internalizing and externalizing behaviors, while distinguishable, may reflect a common neurobiological substrate that results in negative emotionality, a transdiagnostic construct that may explain genetic or Gene × Environment influences on behavior (Rhee, Lahey, & Waldman, Reference Rhee, Lahey and Waldman2015). Many of the conditions we reviewed here result in changes in internalizing and externalizing behaviors, or in the case of prenatal alcohol exposure, ID, and IUGR, executive functioning. Executive functions include updating/working memory, inhibition, and shifting/selective attention and may encompass internalizing and externalizing behaviors (Alvarez & Emory, Reference Alvarez and Emory2006; Miyake & Friedman, Reference Miyake and Friedman2012). Executive functioning impairments are associated with most forms of psychopathology and may be a common underlying factor accounting for life span functional impairment, family history of mental illness, prospective psychopathology, worse developmental histories, and more compromised early life brain function (Caspi et al., Reference Caspi, Houts, Belsky, Goldman-Mellor, Harrington, Israel and Moffitt2014; McTeague, Goodkind, & Etkin, Reference McTeague, Goodkind and Etkin2016; Snyder, Miyake, & Hankin, Reference Snyder, Miyake and Hankin2015). A recent study showed that in depression, antidepressant treatment did not result in improvements in executive functioning, suggesting that common treatments are ineffective in affecting this domain (Shilyansky et al., Reference Shilyansky, Williams, Gyurak, Harris, Usherwood and Etkin2016). The conditions we have reviewed here may provide insight into neurobiological substrates that underlie transdiagnostic entities such as negative emotionality or executive functioning, which in turn, may point toward discovery of “generalist” genes (affected by epigenetic mechanisms) and brain structures for targeted therapeutics.
In this article, we have presented information that provides biological plausibility to support the hypothesis that common fetal events affect brain development and function across the life span through a variety of mechanisms. While the literature has moved over the decades from describing the global effects of these events to documenting more regionally specific effects, it still tends to focus on “one event–one brain region” types of analyses. This approach stems from classic neuroscience research thinking in relatively static adult brains where accidents of nature in humans are used to decipher what a given brain area “does” (e.g., a stroke in the motor cortex leads to hemiplegia). The same rules do not appear to apply to the developing fetal and neonatal brain because of the incompleteness of its circuitry at the time of the environmental event and the enormous plasticity that the young brain exhibits. Thus, development itself becomes a mediating factor in the ultimate behavioral outcome.
A greater understanding of how affected (and nonaffected) regions interact at the circuit level is critical to truly understand the effect of prenatal events on complex behaviors. Given the commonality of circuits affected (by global metabolic deficits), we no longer expect to find a “signature” effect on the developing brain based on etiology, but rather based on timing, caused by unbalanced effects on circuit inputs. Thus, different events that have a common timing (e.g., last trimester) might result in unifinality, whereas a single type of environmental disruptor can lead to multiple outcomes (multifinality) based on the timing of that single disruptor (e.g., last trimester vs. toddlerhood for iron).


