


Introduction
Autoimmune disorders are a common source of illness and are often of uncertain origin. The focus of this article is a variant of chronic immune demyelinating polyradiculoneuropathy (CIDP) known as multifocal acquired demyelinating sensory and motor neuropathy (MADSAM) or Lewis–Sumner syndrome (LSS). First described by Lewis et al.Reference Lewis, Sumner, Brown and Asbury1 LSS is estimated to account for 16%–18% of CIDP.Reference Viala, Renie and Maisonobe2, Reference Gorson, Ropper and Weinberg3 It typically presents in the upper limbs and can have either sensory or motor components, such as pain or weakness in discrete areas predicated by peripheral nerve territories. Effective treatments include intravenous immunoglobulin (IVIg), plasmapheresis, and, to an extent, steroids.
The literature on this condition is relatively sparse, many reports being of small patient populations. Another contributing factor to our gap in knowledge is that most of these case studies are limited in detail. Rayjaballi et al. wrote a detailed review of eight cases and reviewed another 82 reported cases that were of pure upper limb onset.Reference Rajabally and Chavada4 However, they were hampered by a lack of available detail and had to include a category for those in whom information was inadequate. Although a review in 2007 by Lewis found over 100 cases of LSS, this is an inadequate sample size for proper scientific inquiry.Reference Lewis5
Diagnostic delay in LSS has the potential for significant ramifications. It has variable presentations so is likely to be missed, which could explain the small number of cases reported in the literature. We have encountered two cases with closely followed symptoms that worsened to a clinically significant degree after a documented trauma. Finally, we discuss what methods we have found to be effective in achieving good symptomatic control and remission in our patient population.
Methods
This project followed the guidelines set out by the Memorial University of Newfoundland Human Research Ethics Association for a case series. Patients with a diagnosis of LSS were identified by Neurologists at the Health Sciences Center, St. John’s Newfoundland. The patients’ charts were then requisitioned and kept in a locked archive room to maintain strict confidentiality. Each patient was assigned a designation that all information gathered were documented under for their clinical presentation and laboratory study results.
For inclusion into the study, the patients needed to meet diagnostic criteria as per guidelines defined by the European Federation of Neurological Societies/Peripheral Nerve Society.Reference Van den Bergh, Hadden and Bouche6
We recorded the patients’ electrodiagnostic findings upon their first presentation to our clinic and gathered their history and physical information in detail where available. The course of the subjects’ illness and response to treatment were then documented and summarized.
Results
The key clinical and electrophysiological elements for all nine patients can be found in Table 1. Seven of the nine patients were male. The age of onset ranged from 33 to 64 years.
Table 1: The sex, onset characteristics, progression, nerve involvement, and requirement of treatment for nine patients with LSS

IVIg, intravenous immunoglobulin; LSS, Lewis–Sumner syndrome; N/A, not applicable.
a Involved nerves includes any clinically significant electrophysiological deficit.
b Effective treatment defined as arresting the progression of symptoms, clinical improvement, or electrophysiological improvement.
Clinical symptoms at first presentation were confined to the arms in six patients and to the legs in two. Symptoms affected at least one arm and one leg in one patient at presentation. Symptoms progressed to involve both arms and legs in three patients with initial arm involvement, and in two patients with initial leg involvement, giving a total of six patients with both arm and leg involvement.
At onset, symptoms were purely motor in three patients, purely sensory in five and sensorimotor in one. Six progressed to manifest sensorimotor findings; three from pure motor and three from pure sensory initial presentations.
Treatment was required for eight of the nine patients, who received at least one infusion of IVIg and four of them also received oral prednisone. Of the seven patients who received IVIg, two became dependent on continuous infusions, the remainder tapered off of IVIg therapy successfully.
One patient was tested for anti-GM1 and found to be negative. Conduction block in sensory nerves was defined by either temporal dispersion of the response or a large drop in amplitude. In two of our cases, some significant physical trauma was temporally associated with an exacerbation of the symptoms of LSS, as follows:
Patient 1
A 33-year-old male presented in 2007 with upper- and lower-limb weakness and patchy sensory loss. A diagnosis of hereditary motor sensory neuropathy had been made elsewhere, despite the absence of any family history of that condition and negative genetic testing. He had previously presented in 1995 with weakness in his right hand progressing to involve his right foot by 2000. His symptoms were clinically stable until 2005 when he was involved in a severe motor vehicle accident (MVA) which caused a cervical acceleration injury and post-traumatic stress disorder. Three days after which he experienced worsening pain in his right hand. Within 1 month, there was objective wasting of intrinsic muscles of his right hand.
The initial wasting and weakness of the muscles in his right hand increased after the MVA and he began to experience significant pain in both of his hands, requiring narcotic analgesia. He was also found to have mild weakness in his limb girdles and moderate to severe weakness in his distal limb muscles. Electrodiagnostic studies showed conduction block in both his right and left ulnar nerves. Further electrodiagnostic results can be found in Table 2.
Table 2: Electrodiagnostic findings of Patient 1 on first presentation to our clinic
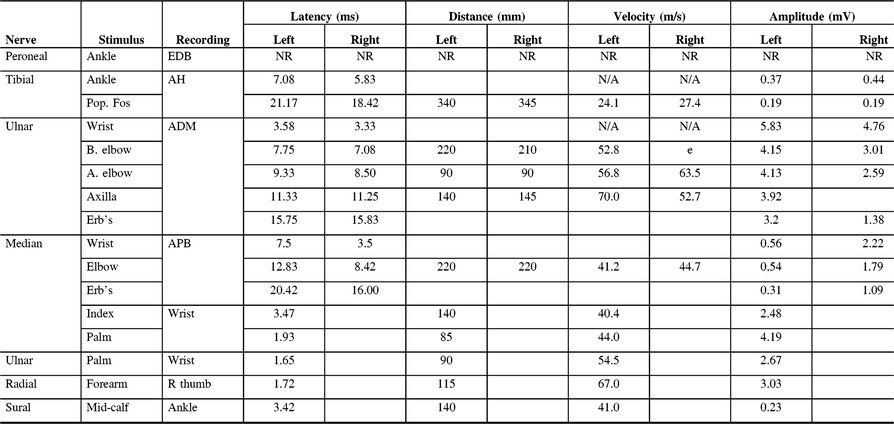
ADM, abductor digiti minimi; AH, abductor hallucis; APB, abductor pollicis brevis; EDB, extensor digitorum brevis; NR, non-reactive.
He also complained of loss of sensation in the feet with a substantial weakness of the ankle dorsiflexors and a steppage gait such that he required several breaks to walk 500 m. The ankle reflexes were absent. Conduction block was later detected in both right and left tibial nerves, left median, and left ulnar nerves. Peroneal motor responses were absent. Neither right nor left peroneal sensory potentials could be recorded.
The asymmetrical sensorimotor involvement with persistent conduction block led to a diagnosis of LSS and IVIg treatment was instituted with monthly infusions over the following 5 years. Sensation returned in his legs although pinprick sensation in his feet only later returned to normal. Almost all measures of power increased significantly, typically by at least one point on the Medical Research Council scale over the next year.
The patient has remained neurologically stable without monthly infusions of IVIg, although there remains significant weakness bilaterally in his median-nerve innervated muscles and a foot slap in his gait for which orthotics are still required. Because of the reduction in pain, he has now ceased taking narcotic analgesics.
Patient 8
This 62-year-old male presented in 2002 with upper limb sensorimotor symptoms and an Adie’s pupil. Initially, he had reported mild, bilateral wasting of the intrinsic muscles of his hands and marked bilateral wasting of the intrinsic muscles of his feet. Electrodiagnostic studies showed absent right ulnar sensory, right peroneal sensory, and right sural sensory nerve potentials. Further electrodiagnostic information can be found in Table 3. Anti-GM1 antibody studies were negative. He remained stable until 2006 when he underwent several consecutive back surgeries. In the weeks following the last of these surgeries, he experienced worsening pain in his right foot and new pain in his entire left leg below the knee which was previously absent. This contributed to a fall that led to a hairline fracture in his foot. His upper limb examination at this time was unremarkable. Follow-up electrodiagnostic studies at this time showed new findings in that the right and left peroneal, left tibial motor, right sural sensory and left sural sensory responses were unobtainable.
Table 3: Electrodiagnostic findings of Patient 8 on first presentation to our clinic
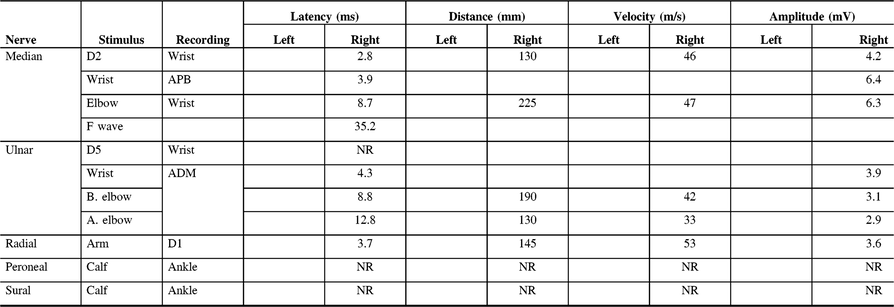
D1, first digit; D2, second digit; D5, fifth digit; ADM, abductor digiti minimi; APB, abductor pollicis brevis; NR, non-reactive.
He was suspected of having LSS and made a good recovery from his symptoms only after starting a regime of prednisone, azathioprine, and IVIG. Neither his Adie’s pupil nor his sensory symptoms fully remitted. He still has some loss of pinprick and light touch sensation in his distal lower limbs. His most recent nerve conduction studies showed unobtainable right peroneal, right tibial, left ulnar sensory, right ulnar sensory, and right sural sensory nerve potentials.
Discussion
The most striking finding in our study population was the temporal association between a physically traumatic event and worsening of the disease. Out of nine patients, there were two who had experienced an exacerbation of symptoms after being exposed to significant trauma. Critically, the exposure to trauma and ensuing worsening of disease process occurred after the patient was already subject to close neurological follow-up. This mitigates the chances of a selection bias, where the illness was already present and was only discovered because a trauma had brought the patient to the hospital. Such events support the hypothesis that trauma may play a role in the autoimmune aspect of the disease. It would be difficult to organize a study with enough power to prove an absolute link between the two given the relative rarity of people involved in traumas who also have an established diagnosis of LSS or other CIDP. However, a similar relationship between trauma and neurologic inflammation is seen in other peripheral nerve conditions.
There is an established link between stress and autoimmune disorders of many different systems.Reference Stojanovich and Marisavljevich7–Reference Kuwabara, Uzawa and Mori14 Although, there are many neurological conditions documented to be triggered or exacerbated by trauma, to our knowledge, this link has not been demonstrated in LSS. An association between surgical intervention and inflammatory nerve injury has, similarly, emerged. Staff et al. described a series of 21 patients who experienced post-operative inflammatory neuropathies.Reference Staff, Engelstad and Klein15 Critically, it did not appear to matter what surgical procedure their patients received, and that they improved on steroid and/or IVIg. More recently, Freo et al. describe the case of a post-operative brachial plexopathy that had objective findings on physical exam and EMG which only improved after administration of IVIg after failing steroids and neuropathic pain agents.Reference Freo, Zara, Furnari and Ori16 Further similarities are reflected in the clinical course of brachial plexitis also known as Parsonage–Turner syndrome (PTS). Unlike in our cases, there is no disease present until after an insult to the body has occurred. However, the relationship between physical trauma and inflammation of the nerves is well documented in PTS. Furthermore, as in our cases of LSS, new cases of PTS have been triggered by a variety of inciting events such as burns, surgery, infection, and endurance exercise.Reference Zhao, Xian and Yu17–Reference Weng and Fidel20 These cases draw many parallels with our own experience, especially that of treating Patient 8 who experienced LSS exacerbation after repeated surgical intervention.
Because of a missed diagnosis, one patient became addicted to the narcotic analgesics that had been used to ease his chronic pain. The damage done here was twofold. The diagnostic delay precluded appropriate treatment so the disease could progress unimpeded; the patient became addicted to narcotic medication, an affliction that had serious ramifications to his personal life. Furthermore, chronic opioid usage has been shown to increase natural cytotoxicityReference Jonsdottir21 and interacts with the Hypothalamus–Pituitary–Adrenal (HPA) axis as well as the regulation of lymphocyte and natural killer cell production.Reference Hall, Suo and Weber22 This may have contributed to this patient’s disease process.
Effective Treatment
Among our nine cases, seven required treatment. The factor that consistently led to positive outcomes for them was access to IVIg therapy. We found IVIg to be superior to prednisone in terms of effectiveness and side effect profile which is in keeping with the literature.Reference Viala, Renie and Maisonobe2 Four of nine patients required indefinite treatment to prevent exacerbations; this is known to happen in some cases.Reference Rajabally and Chavada4, Reference Attarian, Verschueren, Franques, Salort-Campana, Jouve and Pouget23 Azathioprine was used with little effect in two patients. Limited effectiveness of steroid-sparing agents is prevalent in the literature on LSS and CIDPReference Viala, Renie and Maisonobe2, Reference Attarian, Verschueren, Franques, Salort-Campana, Jouve and Pouget23–Reference Sederholm25 but it is not shown that they perform better than placebo level in a randomized control trial. In some cases where patients with CIDP had been noted to improve while taking azathioprine, they were either taking concomitant steroids or only experienced mild amelioration of some symptoms but with exacerbations of others.Reference Thomas, Walker and Rudge26 While this is not a recommendation against the use of azathioprine, it would be prudent to reserve the use of this agent as an option when other treatments are ineffective or not tolerated.
Limitations
Because this study represents a chart review, there are some gaps present in the patients’ histories. Furthermore, when nerve conduction studies were being performed, research was not the primary aim. It is possible that electrodiagnostic findings among our patients could have been more extensive than what was recorded in the EMG laboratory. Many of the patients’ charts are from a time when anti-GM1 testing was not available at our institution. To repeat the testing now would not be practical and so it is not available data for many of our patients.
Acknowledgments
The authors are grateful to Dr. William Pryse-Phillips for providing mentorship throughout the project and Dr. Mark Stefanelli for contributing available cases of LSS. We would also like to acknowledge the late Dr. Richard Marceau for helping to procure funding without which this project may not have been completed. We would also like to thank Ms. Charlene Bowder and Mrs. Patricia Kelly for their administrative assistance and the Seeds, Bridging, and Multidisciplinary fund for supporting the project financially.
Statement of Authorship
Isaac Hughes was involved in conception of the work, data collection, data analysis and interpretation, drafting the article, critical revisions of the article, and final approval of the version to be published.
Alan Goodridge was involved in the conception of the work, data analysis and interpretation, critical revisions of the article, and final approval of the version to be published.
Disclosures
Isaac Hughes was employed by Alan Goodridge and paid with funding provided by the Seeds, Bridging, and Multidisciplinary fund at Memorial University of Newfoundland. Otherwise, neither author has any conflict of interest to disclose.



